

Integrating influenza antigenic dynamics with molecular evolution
- PMID: 24497547
- PMCID: PMC3909918
- DOI: 10.7554/eLife.01914
Integrating influenza antigenic dynamics with molecular evolution
Abstract
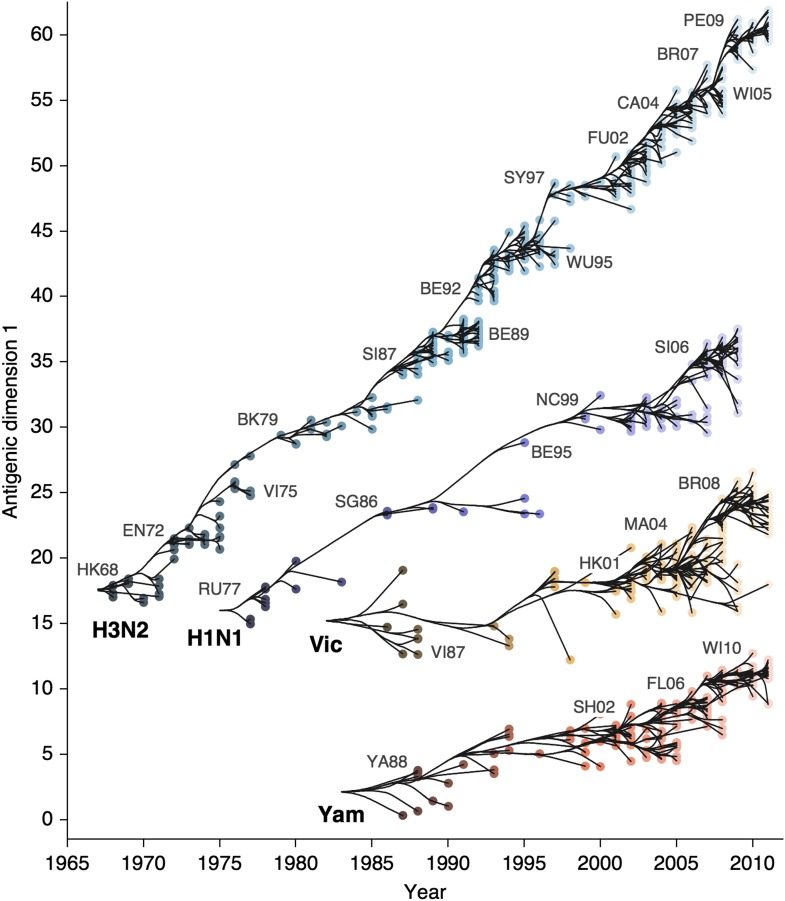
Influenza viruses undergo continual antigenic evolution allowing mutant viruses to evade host immunity acquired to previous virus strains. Antigenic phenotype is often assessed through pairwise measurement of cross-reactivity between influenza strains using the hemagglutination inhibition (HI) assay. Here, we extend previous approaches to antigenic cartography, and simultaneously characterize antigenic and genetic evolution by modeling the diffusion of antigenic phenotype over a shared virus phylogeny. Using HI data from influenza lineages A/H3N2, A/H1N1, B/Victoria and B/Yamagata, we determine patterns of antigenic drift across viral lineages, showing that A/H3N2 evolves faster and in a more punctuated fashion than other influenza lineages. We also show that year-to-year antigenic drift appears to drive incidence patterns within each influenza lineage. This work makes possible substantial future advances in investigating the dynamics of influenza and other antigenically-variable pathogens by providing a model that intimately combines molecular and antigenic evolution. DOI: http://dx.doi.org/10.7554/eLife.01914.001.
Keywords: Bayesian inference; antigenic cartography; evolution; influenza; multidimensional scaling; phylogenetics.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures












References
-
- Ansaldi F, D’Agaro P, de Florentiis D, Puzelli S, Lin YP, Gregory V, Bennett M, Donatelli I, Gasparini R, Crovari P, Hay A, Campello C. 2003. Molecular characterization of influenza B viruses circulating in northern Italy during the 2001–2002 epidemic season. Journal of Medical Virology 70:463–469. 10.1002/jmv.10418 - DOI - PubMed
-
- Ansaldi F, Bacilieri S, Amicizia D, Valle L, Banfi F, Durando P, Sticchi L, Gasparini R, Icardi G, Crovari P. 2004. Antigenic characterisation of influenza B virus with a new microneutralisation assay: comparison to haemagglutination and sequence analysis. Journal of Medical Virology 74:141–146. 10.1002/jmv.20157 - DOI - PubMed
-
- Barr I, Komadina N, Durrant C, Sjogren H, Hurt A, Shaw RP. 2006. Circulation and antigenic drift in human influenza B viruses in SE Asia and Oceania since 2000. Communicable diseases intelligence 30:350–357 - PubMed
-
- Barr I McCauley J Cox N Daniels R Engelhardt O Fukuda K Grohmann G Hay A Kelso A Klimov A Odagiri T Smith D Russell C Tashiro M Webby R Wood J Ye Z Zhang W, Writing Committee of the World Health Organization Consultation on Northern Hemisphere Influenza Vaccine Composition for 2009-2010 . 2010. Epidemiological, antigenic and genetic characteristics of seasonal influenza A (H1N1), A (H3N2) and B influenza viruses: basis for the WHO recommendation on the composition of influenza vaccines for use in the 2009-2010 Northern Hemisphere season. Vaccine 28:1156–1167. 10.1016/j.vaccine.2009.11.043 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
