

Single cell genomics: advances and future perspectives
- PMID: 24497842
- PMCID: PMC3907301
- DOI: 10.1371/journal.pgen.1004126
Single cell genomics: advances and future perspectives
Abstract
Advances in whole-genome and whole-transcriptome amplification have permitted the sequencing of the minute amounts of DNA and RNA present in a single cell, offering a window into the extent and nature of genomic and transcriptomic heterogeneity which occurs in both normal development and disease. Single-cell approaches stand poised to revolutionise our capacity to understand the scale of genomic, epigenomic, and transcriptomic diversity that occurs during the lifetime of an individual organism. Here, we review the major technological and biological breakthroughs achieved, describe the remaining challenges to overcome, and provide a glimpse into the promise of recent and future developments.
Conflict of interest statement
I have read the journal's policy and have the following conflicts: TV is a named inventor on submitted patent applications PCT/EP2011/060211; PCT/EP2013/070858; ZL913096.
Figures
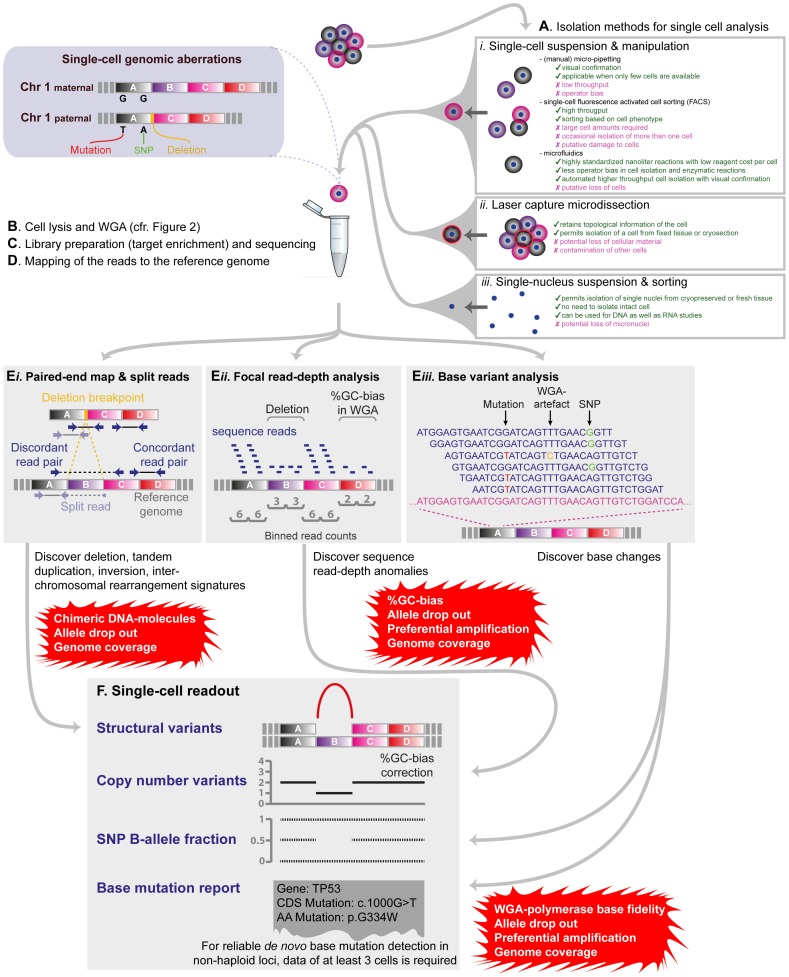
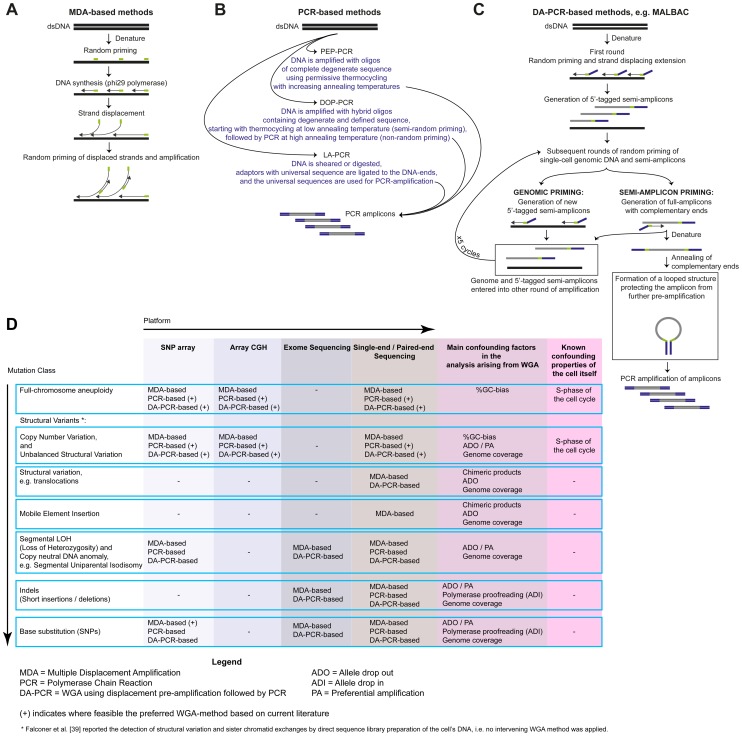
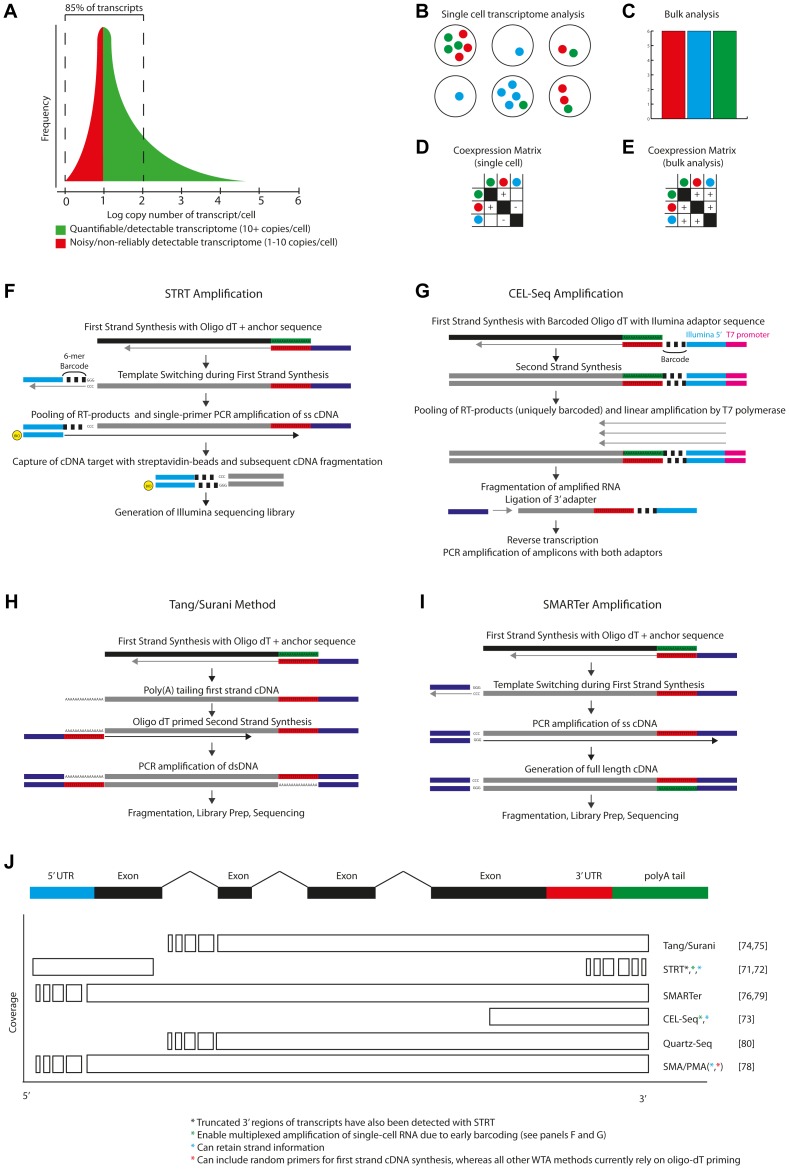
References
-
- Nussbaum RL, McInnes RR, Willard HF (2007) Thompson & Thompson Genetics in Medicine. Philadelphia: Saunders Elsevier. 600 p.
-
- Strachan T, Andrew R (2010) Human Molecular Genetics. New York: Garland Science. 781 p.
-
- Biesecker LG, Spinner NB (2013) A genomic view of mosaicism and human disease. Nat Rev Genet 14: 307–320. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
