

Screening of CACNA1A and ATP1A2 genes in hemiplegic migraine: clinical, genetic, and functional studies
- PMID: 24498617
- PMCID: PMC3865589
- DOI: 10.1002/mgg3.24
Screening of CACNA1A and ATP1A2 genes in hemiplegic migraine: clinical, genetic, and functional studies
Abstract
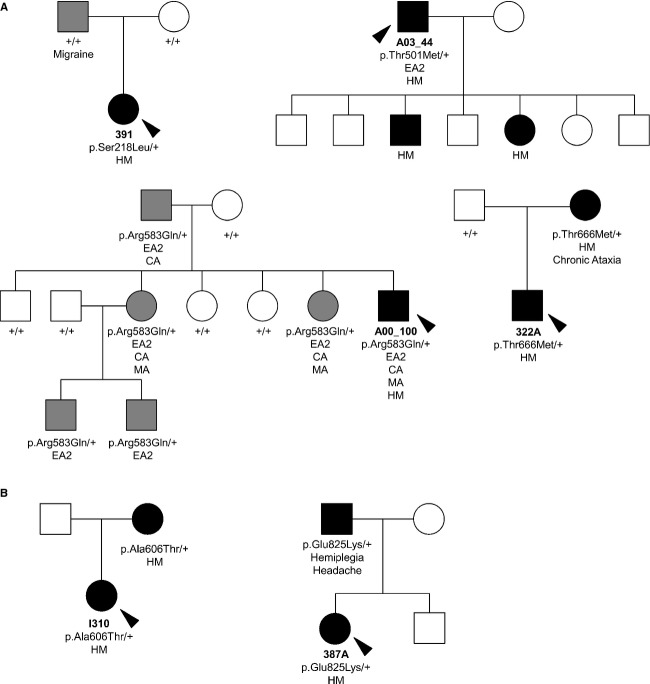
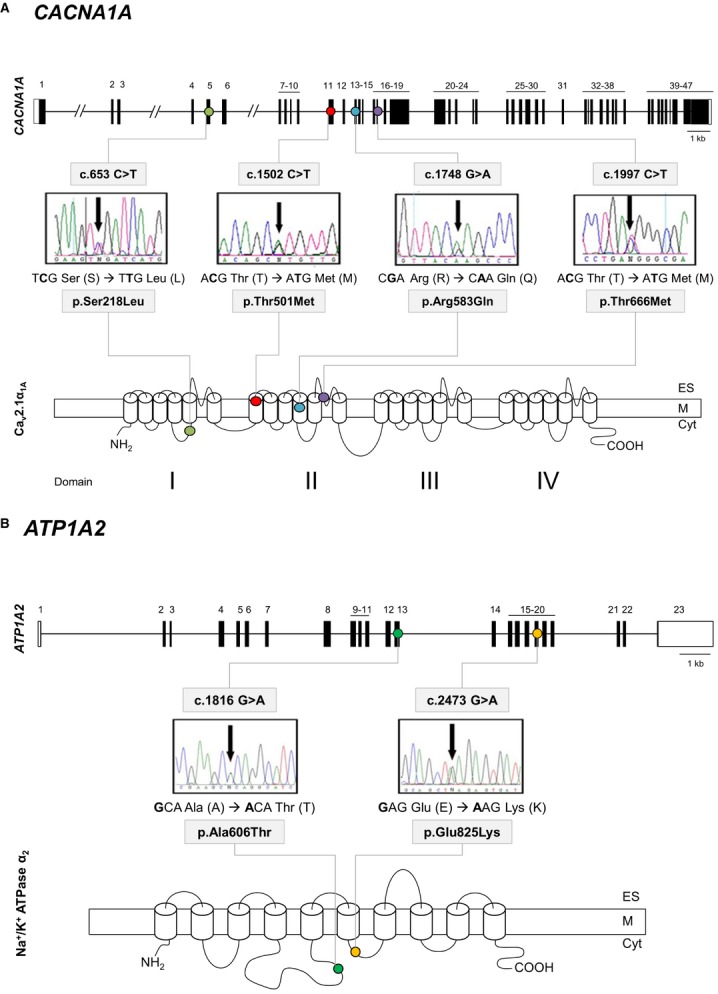
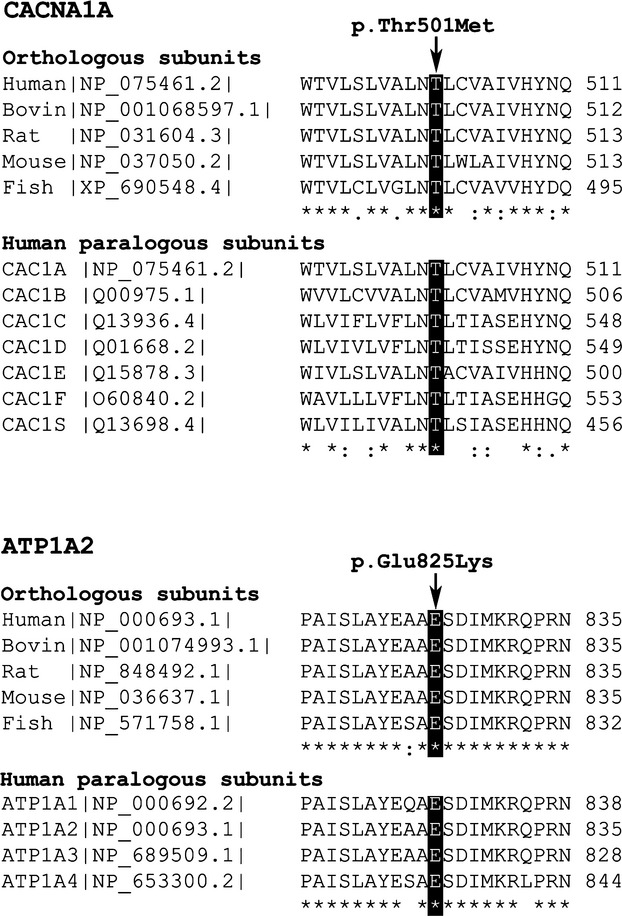
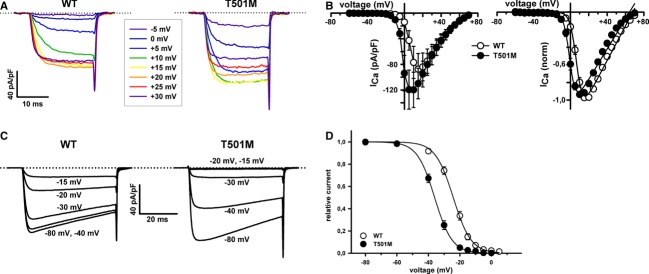
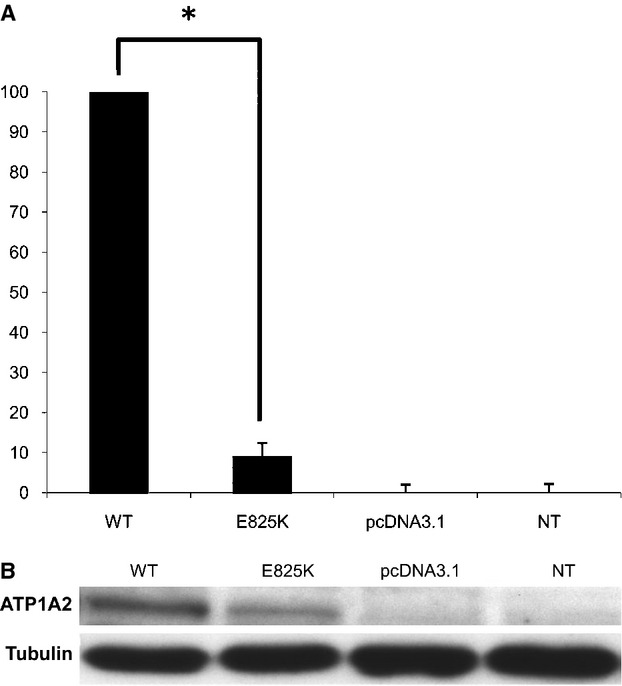
Hemiplegic migraine (HM) is a rare and severe subtype of autosomal dominant migraine, characterized by a complex aura including some degree of motor weakness. Mutations in four genes (CACNA1A, ATP1A2, SCN1A and PRRT2) have been detected in familial and in sporadic cases. This genetically and clinically heterogeneous disorder is often accompanied by permanent ataxia, epileptic seizures, mental retardation, and chronic progressive cerebellar atrophy. Here we report a mutation screening in the CACNA1A and ATP1A2 genes in 18 patients with HM. Furthermore, intragenic copy number variant (CNV) analysis was performed in CACNA1A using quantitative approaches. We identified four previously described missense CACNA1A mutations (p.Ser218Leu, p.Thr501Met, p.Arg583Gln, and p.Thr666Met) and two missense changes in the ATP1A2 gene, the previously described p.Ala606Thr and the novel variant p.Glu825Lys. No structural variants were found. This genetic screening allowed the identification of more than 30% of the disease alleles, all present in a heterozygous state. Functional consequences of the CACNA1A-p.Thr501Met mutation, previously described only in association with episodic ataxia, and ATP1A2-p.Glu825Lys, were investigated by means of electrophysiological studies, cell viability assays or Western blot analysis. Our data suggest that both these variants are disease-causing.
Keywords: ATP1A2; CACNA1A; functional studies; hemiplegic migraine; mutation analysis.
Figures
References
-
- Alonso I, Barros J, Tuna A, Coelho J, Sequeiros J, Silveira I, et al. Phenotypes of spinocerebellar ataxia type 6 and familial hemiplegic migraine caused by a unique CACNA1A missense mutation in patients from a large family. Arch. Neurol. 2003;60:610–614. - PubMed
-
- Battistini S, Stenirri S, Piatti M, Gelfi C, Righetti PG, Rocchi R, et al. A new CACNA1A gene mutation in acetazolamide-responsive familial hemiplegic migraine and ataxia. Neurology. 1999;53:38–43. - PubMed
-
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat. Med. 2002;8:136–142. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
