

Calcium inhibition of ribonuclease H1 two-metal ion catalysis
- PMID: 24499076
- PMCID: PMC3985467
- DOI: 10.1021/ja411408x
Calcium inhibition of ribonuclease H1 two-metal ion catalysis
Abstract
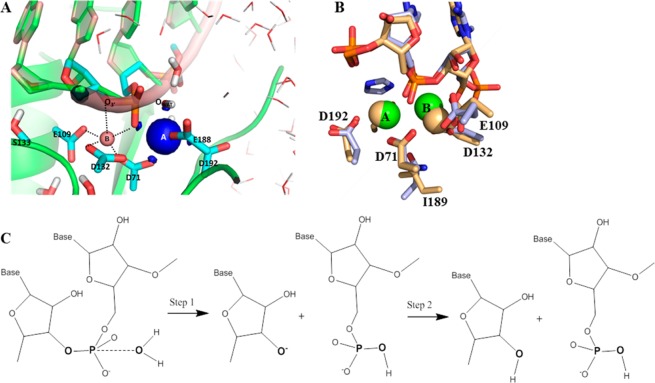
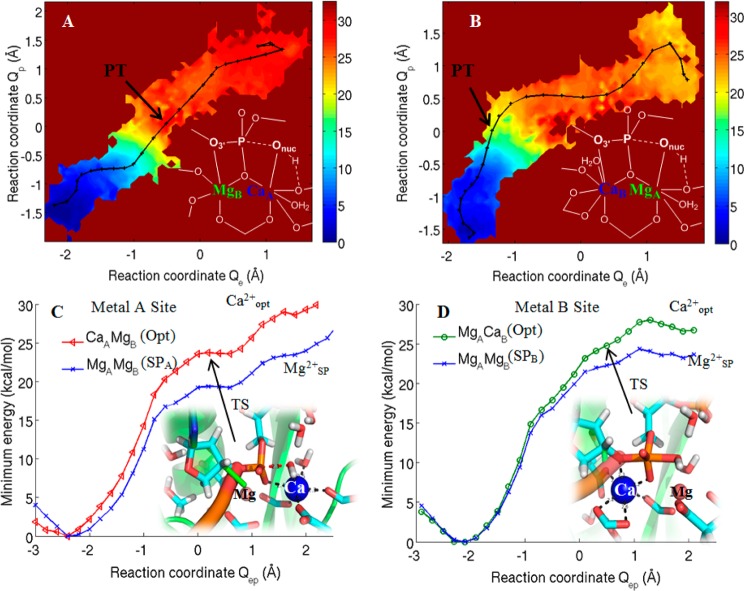
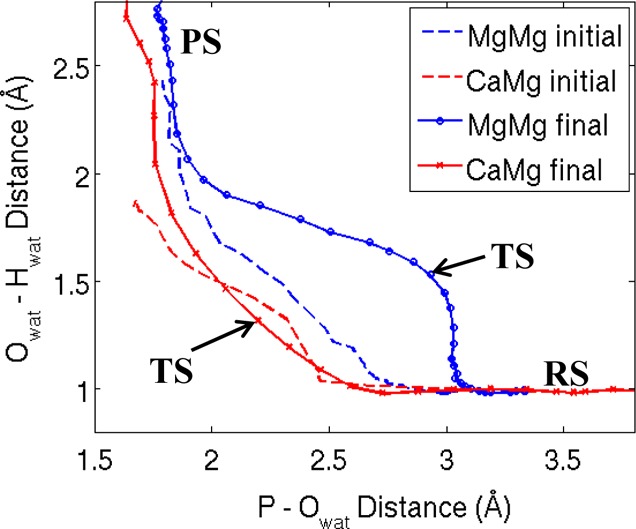
Most phosphate-processing enzymes require Mg(2+) as a cofactor to catalyze nucleotide cleavage and transfer reactions. Ca(2+) ions inhibit many of these enzymatic activities, despite Ca(2+) and Mg(2+) having comparable binding affinities and overall biological abundances. Here we study the molecular details of the calcium inhibition mechanism for phosphodiester cleavage, an essential reaction in the metabolism of nucleic acids and nucleotides, by comparing Ca(2+)- and Mg(2+) catalyzed reactions. We study the functional roles of the specific metal ion sites A and B in enabling the catalytic cleavage of an RNA/DNA hybrid substrate by B. halodurans ribonuclease (RNase) H1 using hybrid quantum-mechanics/molecular mechanics (QM/MM) free energy calculations. We find that Ca(2+) substitution of either of the two active-site Mg(2+) ions substantially increases the height of the reaction barrier and thereby abolishes the catalytic activity. Remarkably, Ca(2+) at the A site is inactive also in Mg(2+)-optimized active-site structures along the reaction path, whereas Mg(2+) substitution recovers activity in Ca(2+)-optimized structures. Geometric changes resulting from Ca(2+) substitution at metal ion site A may thus be a secondary factor in the loss of catalytic activity. By contrast, at metal ion site B geometry plays a more important role, with only a partial recovery of activity after Mg(2+) substitution in Ca(2+)-optimized structures. Ca(2+)-substitution also leads to a change in mechanism, with deprotonation of the water nucleophile requiring a closer approach to the scissile phosphate, which in turn increases the barrier. As a result, Ca(2+) is less efficient in activating the water. As a likely cause for the different reactivities of Mg(2+) and Ca(2+) ions in site A, we identify differences in charge transfer to the ions and the associated decrease in the pKa of the oxygen nucleophile attacking the phosphate group.
Figures
Similar articles
-
Phosphodiester cleavage in ribonuclease H occurs via an associative two-metal-aided catalytic mechanism.J Am Chem Soc. 2008 Aug 20;130(33):10955-62. doi: 10.1021/ja8005786. Epub 2008 Jul 29. J Am Chem Soc. 2008. PMID: 18662000 Free PMC article.
-
Understanding the effect of magnesium ion concentration on the catalytic activity of ribonuclease H through computation: does a third metal binding site modulate endonuclease catalysis?J Am Chem Soc. 2010 Oct 6;132(39):13702-12. doi: 10.1021/ja102933y. J Am Chem Soc. 2010. PMID: 20731347 Free PMC article.
-
Catalytic mechanism of RNA backbone cleavage by ribonuclease H from quantum mechanics/molecular mechanics simulations.J Am Chem Soc. 2011 Jun 15;133(23):8934-41. doi: 10.1021/ja200173a. Epub 2011 May 24. J Am Chem Soc. 2011. PMID: 21539371 Free PMC article.
-
Catalytic metal ions and enzymatic processing of DNA and RNA.Acc Chem Res. 2015 Feb 17;48(2):220-8. doi: 10.1021/ar500314j. Epub 2015 Jan 15. Acc Chem Res. 2015. PMID: 25590654 Review.
-
Catalytic strategies of self-cleaving ribozymes.Acc Chem Res. 2008 Aug;41(8):1027-35. doi: 10.1021/ar800050c. Epub 2008 Jul 25. Acc Chem Res. 2008. PMID: 18652494 Review.
Cited by
-
The control of the discrimination between dNTP and rNTP in DNA and RNA polymerase.Proteins. 2016 Nov;84(11):1616-1624. doi: 10.1002/prot.25104. Epub 2016 Aug 10. Proteins. 2016. PMID: 27480935 Free PMC article.
-
Structure of HIV-1 reverse transcriptase cleaving RNA in an RNA/DNA hybrid.Proc Natl Acad Sci U S A. 2018 Jan 16;115(3):507-512. doi: 10.1073/pnas.1719746115. Epub 2018 Jan 2. Proc Natl Acad Sci U S A. 2018. PMID: 29295939 Free PMC article.
-
Not making the cut: Techniques to prevent RNA cleavage in structural studies of RNase-RNA complexes.J Struct Biol X. 2022 Mar 11;6:100066. doi: 10.1016/j.yjsbx.2022.100066. eCollection 2022. J Struct Biol X. 2022. PMID: 35340590 Free PMC article. Review.
-
Dynamic coordination of two-metal-ions orchestrates λ-exonuclease catalysis.Nat Commun. 2018 Oct 23;9(1):4404. doi: 10.1038/s41467-018-06750-9. Nat Commun. 2018. PMID: 30353000 Free PMC article.
-
Fibrillarin Ribonuclease Activity is Dependent on the GAR Domain and Modulated by Phospholipids.Cells. 2020 May 6;9(5):1143. doi: 10.3390/cells9051143. Cells. 2020. PMID: 32384686 Free PMC article.
References
-
- Stahley M. R.; Strobel S. A. Science 2005, 309, 1587. - PubMed
-
- Sträter N.; Lipscomb W. N.; Klabunde T.; Krebs B. Angew. Chem., Int. Ed. 1996, 35, 2024.
-
- Yang W.; Lee J. Y.; Nowotny M. Mol. Cell 2006, 22, 5. - PubMed
-
- Gerlt J. A.; Coderre J. A.; Mehdi S. Adv. Enzymol. Relat. Areas Mol. Biol. 1983, 55, 291. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
