

Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes
- PMID: 24501229
- PMCID: PMC3963529
- DOI: 10.1136/jmedgenet-2013-102116
Genome-wide DNA methylation analysis of patients with imprinting disorders identifies differentially methylated regions associated with novel candidate imprinted genes
Erratum in
- J Med Genet. 2014 Jul;51(7):478
Abstract
Background: Genomic imprinting is allelic restriction of gene expression potential depending on parent of origin, maintained by epigenetic mechanisms including parent of origin-specific DNA methylation. Among approximately 70 known imprinted genes are some causing disorders affecting growth, metabolism and cancer predisposition. Some imprinting disorder patients have hypomethylation of several imprinted loci (HIL) throughout the genome and may have atypically severe clinical features. Here we used array analysis in HIL patients to define patterns of aberrant methylation throughout the genome.
Design: We developed a novel informatic pipeline capable of small sample number analysis, and profiled 10 HIL patients with two clinical presentations (Beckwith-Wiedemann syndrome and neonatal diabetes) using the Illumina Infinium Human Methylation450 BeadChip array to identify candidate imprinted regions. We used robust statistical criteria to quantify DNA methylation.
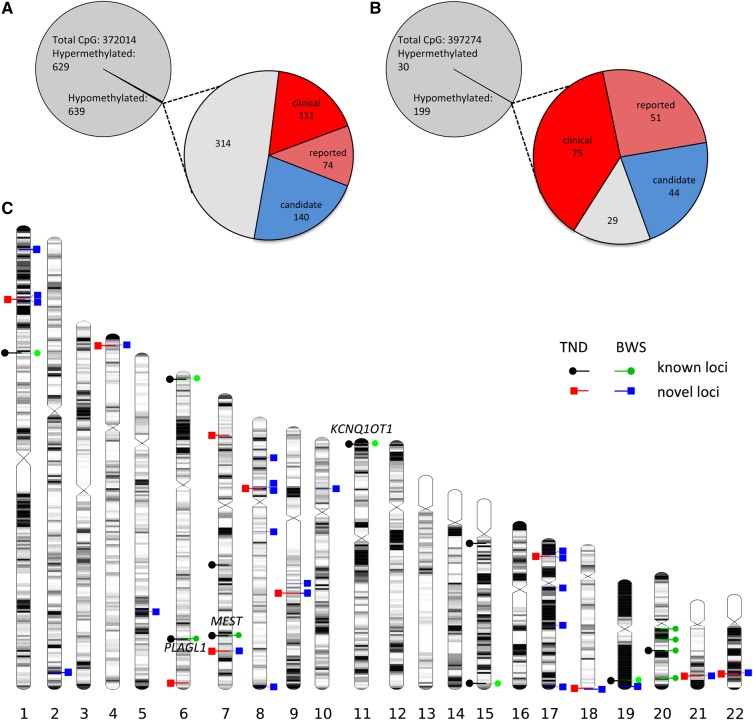
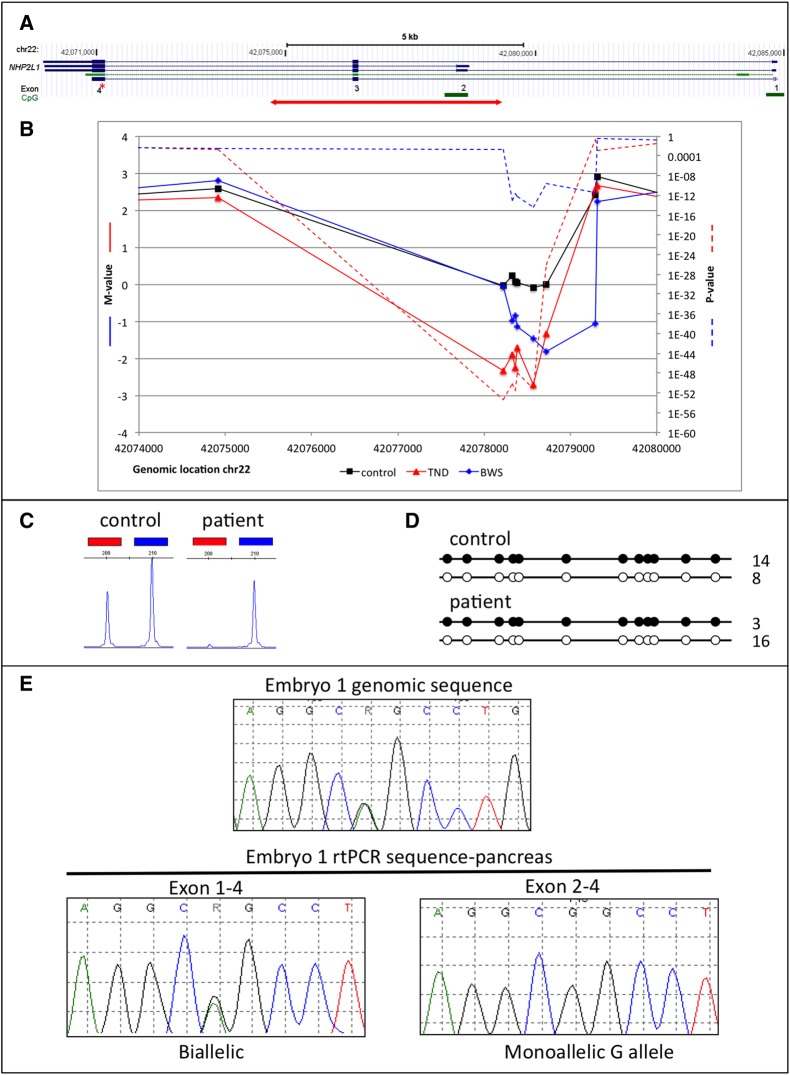
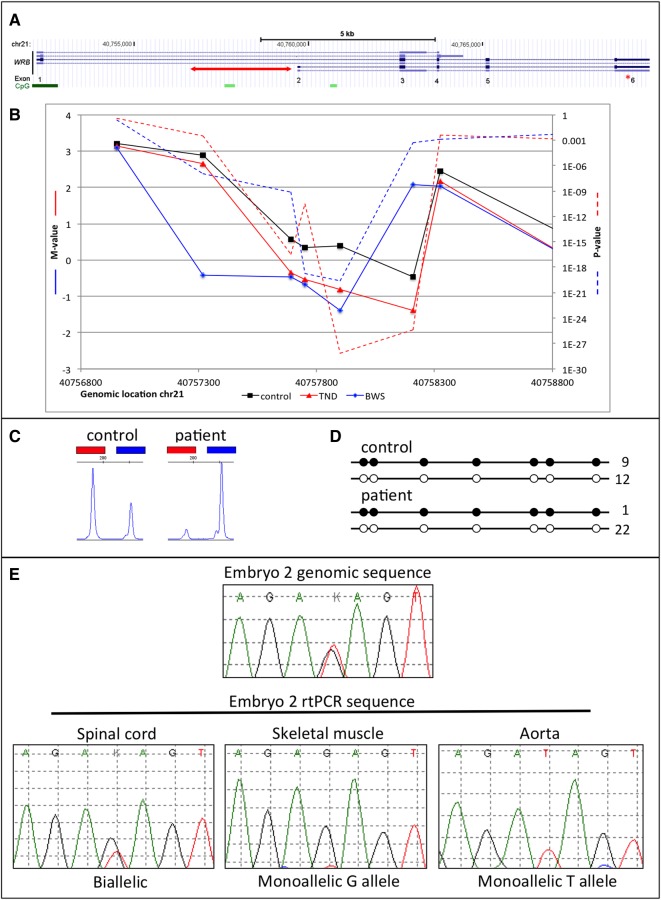
Results: We detected hypomethylation at known imprinted loci, and 25 further candidate imprinted regions (nine shared between patient groups) including one in the Down syndrome critical region (WRB) and another previously associated with bipolar disorder (PPIEL). Targeted analysis of three candidate regions (NHP2L1, WRB and PPIEL) showed allelic expression, methylation patterns consistent with allelic maternal methylation and frequent hypomethylation among an additional cohort of HIL patients, including six with Silver-Russell syndrome presentations and one with pseudohypoparathyroidism 1B.
Conclusions: This study identified novel candidate imprinted genes, revealed remarkable epigenetic convergence among clinically divergent patients, and highlights the potential of epigenomic profiling to expand our understanding of the normal methylome and its disruption in human disease.
Keywords: Epigenetics; Genome-wide; Imprinting.
Figures
Similar articles
-
Methylation screening of reciprocal genome-wide UPDs identifies novel human-specific imprinted genes.Hum Mol Genet. 2011 Aug 15;20(16):3188-97. doi: 10.1093/hmg/ddr224. Epub 2011 May 18. Hum Mol Genet. 2011. PMID: 21593219
-
Genome-wide allelic methylation analysis reveals disease-specific susceptibility to multiple methylation defects in imprinting syndromes.Hum Mutat. 2013 Apr;34(4):595-602. doi: 10.1002/humu.22276. Epub 2013 Feb 19. Hum Mutat. 2013. PMID: 23335487
-
Genome-wide survey of parent-of-origin effects on DNA methylation identifies candidate imprinted loci in humans.Hum Mol Genet. 2018 Aug 15;27(16):2927-2939. doi: 10.1093/hmg/ddy206. Hum Mol Genet. 2018. PMID: 29860447 Free PMC article.
-
Epigenetic and genetic alterations of the imprinting disorder Beckwith-Wiedemann syndrome and related disorders.J Hum Genet. 2013 Jul;58(7):402-9. doi: 10.1038/jhg.2013.51. Epub 2013 May 30. J Hum Genet. 2013. PMID: 23719190 Review.
-
Genomic imprinting syndromes and cancer.Adv Genet. 2010;70:145-75. doi: 10.1016/B978-0-12-380866-0.60006-X. Adv Genet. 2010. PMID: 20920748 Review.
Cited by
-
Genome-wide DNA methylation analysis of transient neonatal diabetes type 1 patients with mutations in ZFP57.BMC Med Genet. 2016 Apr 14;17:29. doi: 10.1186/s12881-016-0292-4. BMC Med Genet. 2016. PMID: 27075368 Free PMC article.
-
Genome-Wide Association Study Identifies a Susceptibility Locus for Comitant Esotropia and Suggests a Parent-of-Origin Effect.Invest Ophthalmol Vis Sci. 2018 Aug 1;59(10):4054-4064. doi: 10.1167/iovs.18-24082. Invest Ophthalmol Vis Sci. 2018. PMID: 30098192 Free PMC article.
-
Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement.Nat Rev Endocrinol. 2018 Aug;14(8):476-500. doi: 10.1038/s41574-018-0042-0. Nat Rev Endocrinol. 2018. PMID: 29959430 Free PMC article. Review.
-
Conception by fertility treatment and offspring deoxyribonucleic acid methylation.Fertil Steril. 2021 Aug;116(2):493-504. doi: 10.1016/j.fertnstert.2021.03.011. Epub 2021 Apr 3. Fertil Steril. 2021. PMID: 33823999 Free PMC article.
-
Inference of putative cell-type-specific imprinted regulatory elements and genes during human neuronal differentiation.Hum Mol Genet. 2023 Jan 13;32(3):402-416. doi: 10.1093/hmg/ddac207. Hum Mol Genet. 2023. PMID: 35994039 Free PMC article.
References
-
- Messerschmidt DM, de Vries W, Ito M, Solter D, Ferguson-Smith A, Knowles BB. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 2012;335:1499–502 - PubMed
-
- Peters J, Beechey C. Identification and characterisation of imprinted genes in the mouse. Brief Funct Genomic Proteomic 2004;2:320–33 - PubMed
-
- Yamazawa K, Ogata T, Ferguson-Smith AC. Uniparental disomy and human disease: an overview. Am J Med Genet 2010;154C:329–34 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources