

Review
doi: 10.1021/cr400459c.
Epub 2014 Feb 6.
Conditionally and transiently disordered proteins: awakening cryptic disorder to regulate protein function
Affiliations
- PMID: 24502763
- PMCID: PMC4090257
- DOI: 10.1021/cr400459c
Item in Clipboard
Review
Conditionally and transiently disordered proteins: awakening cryptic disorder to regulate protein function
Chem Rev.
.
No abstract available
Figures
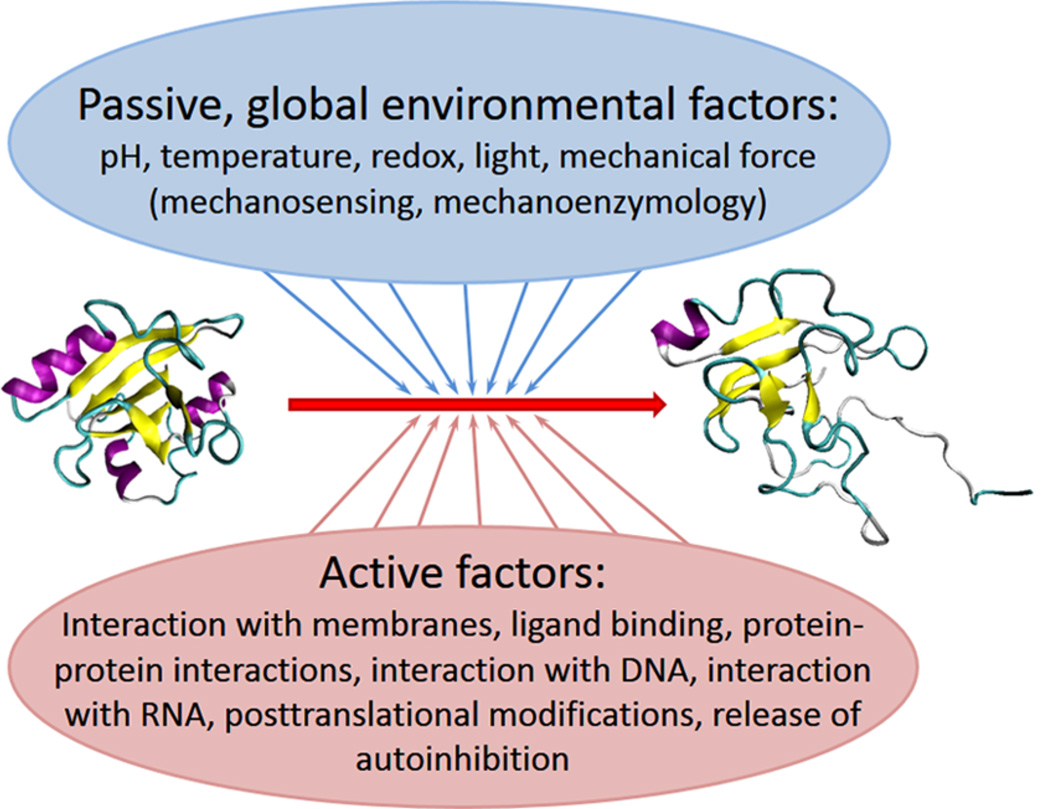
Molecular mechanisms underlying functional unfolding of proteins. An ordered (inactive) protein can become active via local or global functional unfolding. This transition can be promoted by passive, global environmental factors (such as changes in pH, temperature increase, changes in the redox potential, application of mechanical force, exposure to light). Alternatively, many active factors (such as interactions with various partners, post-translational modifications, and release of autoinhibition) can also result in functional unfolding.
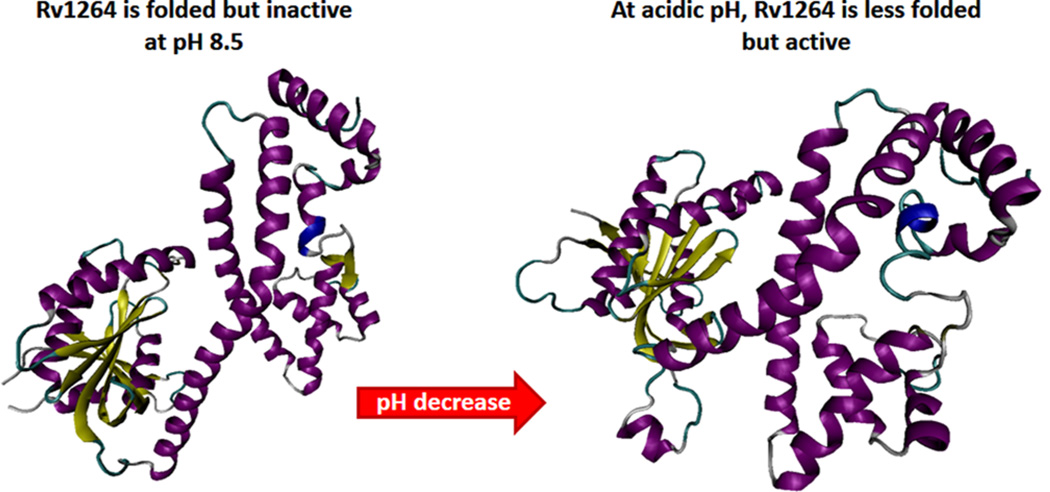
Acid-induced local unfolding leading to activation. Overall structure of Rv1264 in the inhibited (left; PDB ID: 1Y10) and active states (right; PDB ID: 1Y11). Although Rv1264 exists as a dimer, structures of inhibited and active monomers are shown for simplicity. The regulatory domains remain essentially unchanged upon enzyme activation, but the interface with the catalytic domains differs substantially. In fact, in the structure of the inhibited state, catalytic and regulatory domains share a large interface involving catalytic residues. In the structure of the active dimer, the catalytic domains rotate by 55° to form two catalytic sites at their interface. Although in the acidic environment the entire molecule undergoes noticeable structural alterations, the most dramatic structural changes occur at the linker region containing the αN10-switch. In the inhibited state, this segment forms a long α-helix, whereas in the active state, this fragment assumes a disordered conformation with a short helical segment loosely connecting regulatory and catalytic domains.
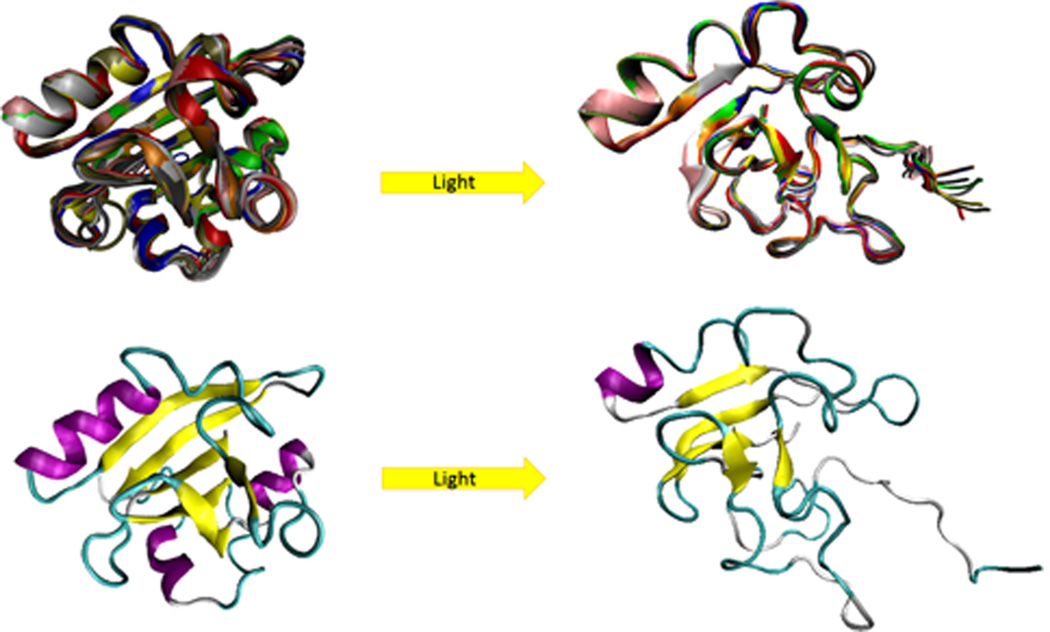
Functional unfolding associated with the light-induced activation of the photoactive yellow protein. Comparison of the ground state (left structure, PDB ID: 3PHY) and the transient light activated signaling state of the photoactive yellow protein (right structure, PDB ID: 2KX6). The top row represents ensembles of these two functional states of the photoactive yellow protein, whereas the bottom row shows representative members of the corresponding ensembles. Ground-state structure was determined by multidimensional NMR spectroscopy, and is in agreement with an earlier published 1.4 Å crystal structure. The light activated state structure was obtained by combining double electron electron resonance spectroscopy (DEER), NMR, and time-resolved pump–probe X-ray solution scattering (TR-SAXS/WAXS) data. It consists of an open, twisted, 6-stranded, antiparallel β-sheet, which is flanked by four α-helices on both sides.,, On the contrary, the light-activated form is highly disordered. This structure satisfies DEER, SAXS/WAXS, and NMR data simultaneously.
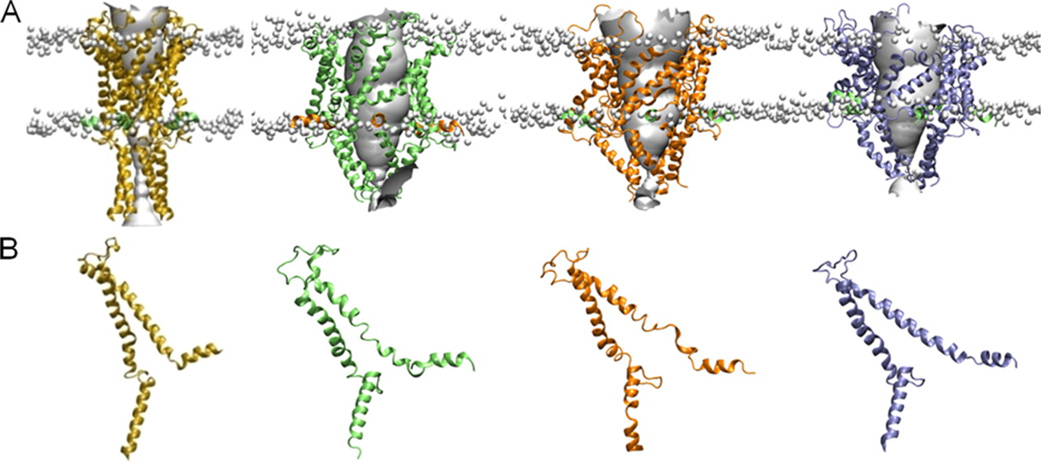
Mechanical activation of the MS channel. Simulation studies incorporating EPR and FRET data. (A) The closed state (yellow) and model open states of the protein obtained under membrane tension of 0 (green), 5 (orange), and 60 mN/m (blue), respectively, viewed in the plane of the membrane. The location of the lipid phosphate head groups is indicated by the gray balls to give an indication of the position of the membrane. (B) The structure of a single subunit of each structure is indicated. Reprinted with permission from ref . Copyright 2010 Rockefeller University Press.
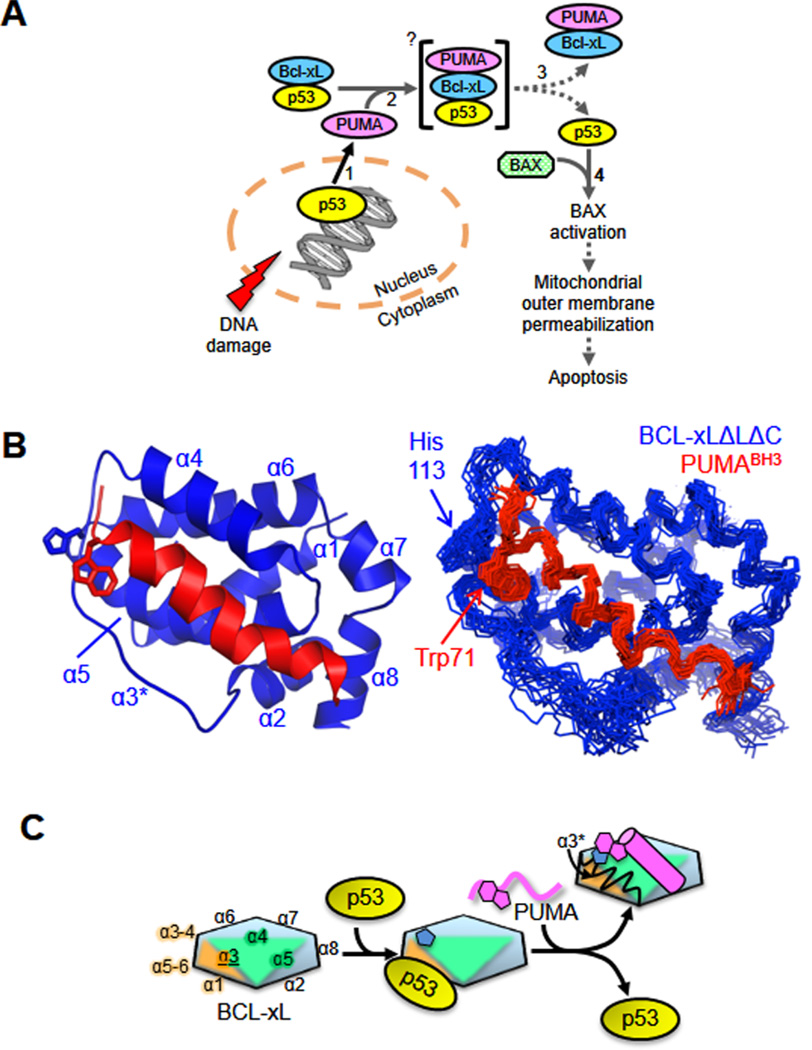
Local unfolding induced by ligand binding. Ligand binding-associated unfolding of two helices within BCL-xL is associated with p53 release and the induction of apoptosis. (A) Scheme illustrating the mechanism by which p53 regulates apoptosis through interactions with DNAin the nucleus and BCL-2 family proteins in the cytosol. Increasing numerals denote the sequence of events involved in this process. (B) Solution structure of the BCL-xLΔLΔC–PUMABH3 complex; ribbon representation of the lowest-energy structure (left) and alignment of the 20 lowest-energy structures (right). The disrupted α3 region is marked with an asterisk. (C) Schematic illustration of the mechanism by which PUMA induces unfolding within α2 and α3 of BCL-xL, which is associated with p53 release. The formation of a π-stacking interaction between His113 of BCL-xL (blue pentagon shapes) and Trp71 of PUMA (magenta geometric shapes) is associated with unfolding of α2 and α3 (α3* in the upper right). BCL-xL is represented as a multicolor hexagon, with the edges representing its α-helices, as marked; PUMA in its unbound form is shown in magenta as a wavy line and as a cylinder when bound to BCL-xL. Adapted with permission from ref . Copyright 2013 Nature Publishing Group.
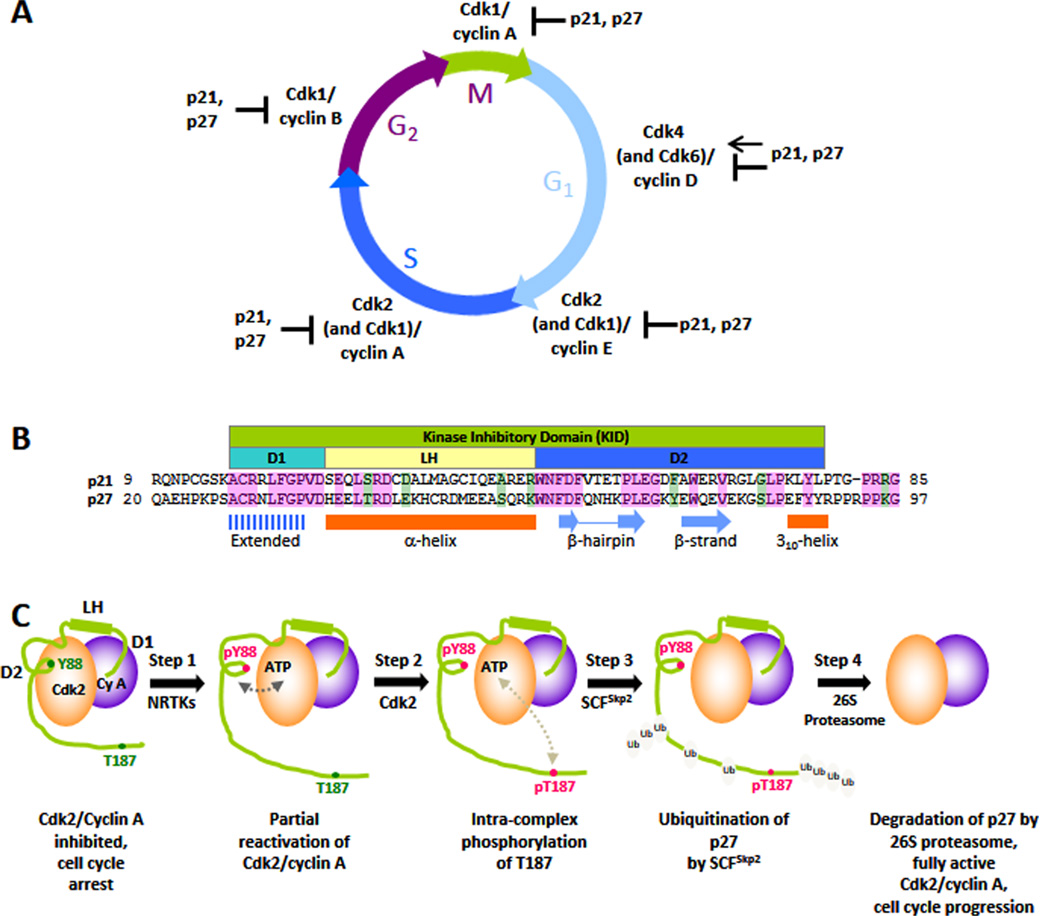
Local functional unfolding promoted by phosphorylation. The disordered protein, p27, is a regulator of eukaryotic cell division (A) and folds upon binding to Cdk/cyclin complexes. The kinase inhibitory domain (B) mediates binding to Cdk/cyclin complexes, while the C-terminal domain (containing T187) remains disordered (C) and plays a critical role in phosphorylation signaling that controls the transition from G1 to S phase of the division cycle. Phosphorylation of Y88 causes unfolding of a structural element that otherwise blocks the binding of ATP to the Cdk2 active site. This so-called “regulated unfolding” reactivates Cdk2, enabling intracomplex phosphorylation of T187 and subsequent ubiquitination and degradation of p27, which fully activates Cdk2 and drives cell cycle progression. Adapted with permission from ref . Copyright 2012 Biochemical Society.
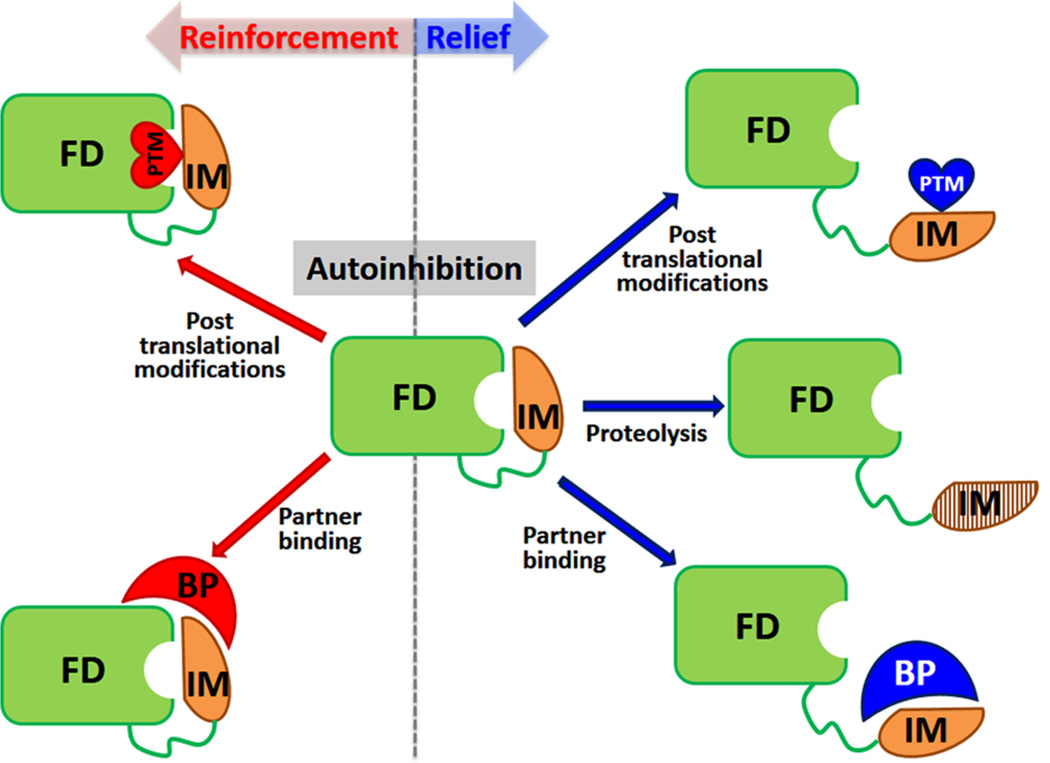
Regulation of autoinhibition. In autoinhibited proteins, an inhibitory module (IM, yellow) modulates/inhibits the activity of a functional domain (FD, green). Equilibrium between active and inhibited states is regulated; that is, autoinhibition can be actively relieved (right side) or reinforced (left side). Reversal of autoinhibition in most systems relies on post-translational modifications (PTMs, hearts, red for reinforcement or blue for relieve), such as phosphorylation, binding to an activating partner (BP, crescent, red for reinforcement and blue for relieve), or proteolytic cleavage of the IM (right side). Reinforcement of autoinhibition has been shown to be caused by binding of partners or PTMs (left side). Autoinhibition can be caused by direct, steric interactions of the IM with the FD but can also be induced allosterically.
References
-
- Fischer E. Ber. Dtsch. Chem. Ges. 1894;27:2985.
-
- Lemieux UR, Spohr U. Adv. Carbohydr. Chem. Biochem. 1994;50:1. - PubMed
-
- Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Genome Inform. Ser. Workshop Genome Inform. 2000;11:161. - PubMed
-
- Vucetic S, Brown CJ, Dunker AK, Obradovic Z. Proteins. 2003;52:573. - PubMed
-
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. J. Mol. Biol. 2004;337:635. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
