

The Pex1-G844D mouse: a model for mild human Zellweger spectrum disorder
- PMID: 24503136
- PMCID: PMC4901203
- DOI: 10.1016/j.ymgme.2014.01.008
The Pex1-G844D mouse: a model for mild human Zellweger spectrum disorder
Abstract
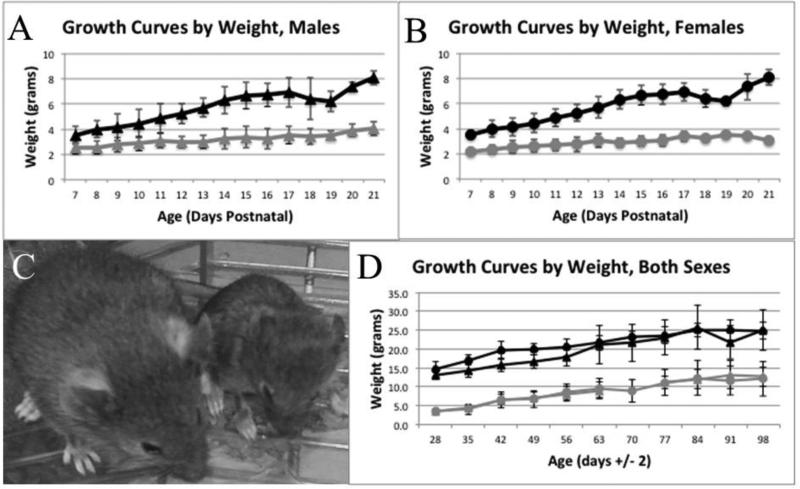
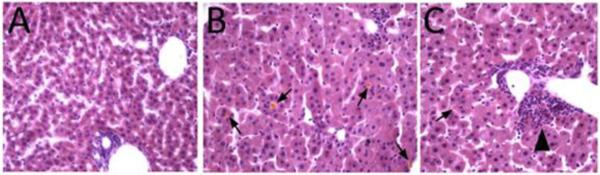
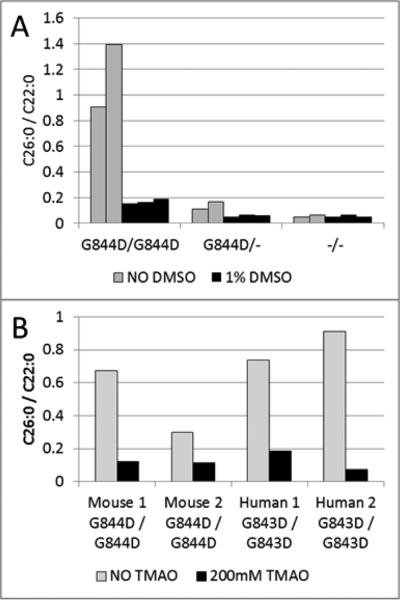
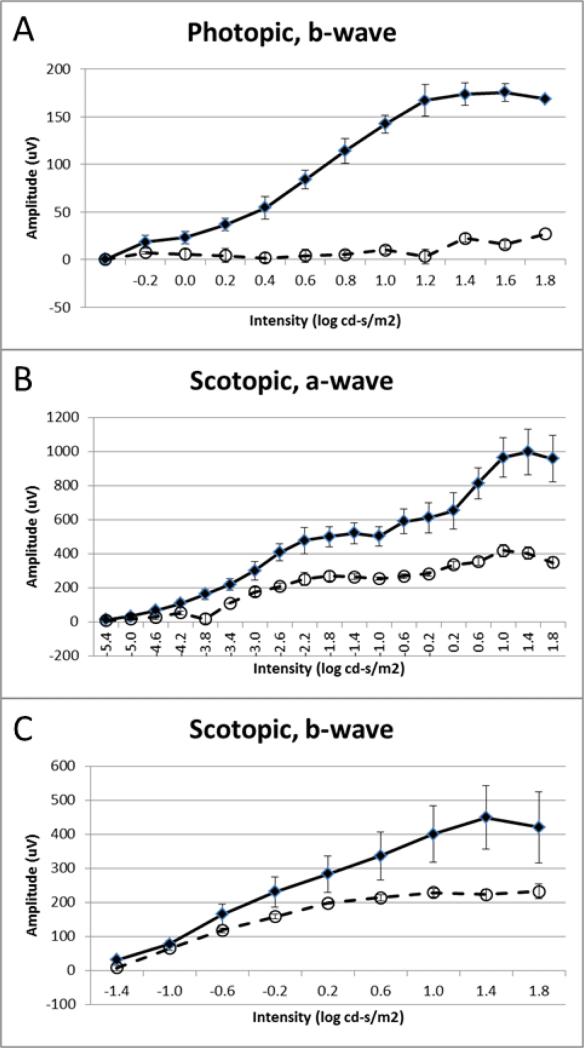
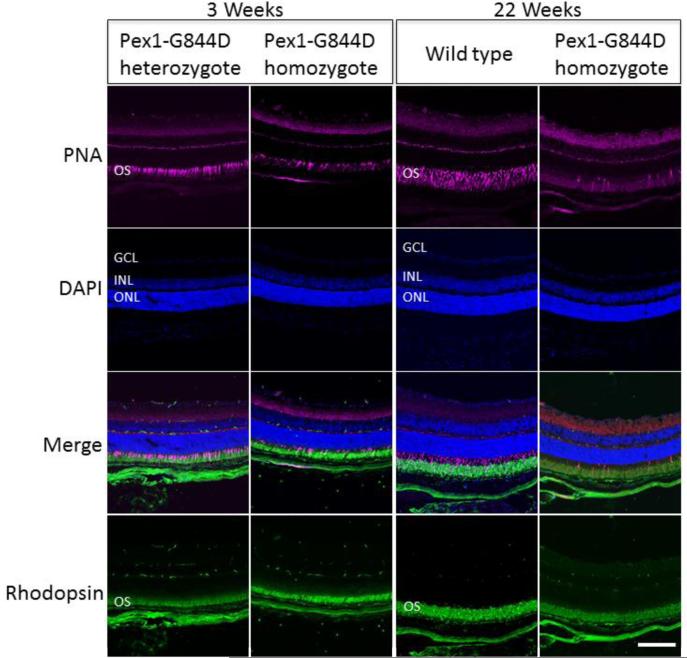
Zellweger spectrum disorder (ZSD) is a disease continuum that results from inherited defects in PEX genes essential for normal peroxisome assembly. These autosomal recessive disorders impact brain development and also cause postnatal liver, adrenal, and kidney dysfunction, as well as loss of vision and hearing. The hypomorphic PEX1-G843D missense allele, observed in approximately 30% of ZSD patients, is associated with milder clinical and biochemical phenotypes, with some homozygous individuals surviving into early adulthood. Nonetheless, affected children with the PEX1-G843D allele have intellectual disability, failure to thrive, and significant sensory deficits. To enhance our ability to test candidate therapies that improve human PEX1-G843D function, we created the novel Pex1-G844D knock-in mouse model that represents the murine equivalent of the common human mutation. We show that Pex1-G844D homozygous mice recapitulate many classic features of mild ZSD cases, including growth retardation and fatty livers with cholestasis. In addition, electrophysiology, histology, and gene expression studies provide evidence that these animals develop a retinopathy similar to that observed in human patients, with evidence of cone photoreceptor cell death. Similar to skin fibroblasts obtained from ZSD patients with a PEX1-G843D allele, we demonstrate that murine cells homozygous for the Pex1-G844D allele respond to chaperone-like compounds, which normalizes peroxisomal β-oxidation. Thus, the Pex1-G844D mouse provides a powerful model system for testing candidate therapies that address the most common genetic cause of ZSD. In addition, this murine model will enhance studies focused on mechanisms of pathogenesis.
Keywords: Bile acids; PEX1; Peroxisome; Photoreceptor degeneration; Retinopathy; Zellweger spectrum disorder.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
References
-
- Wanders RJ, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annual review of biochemistry. 2006;75:295–332. - PubMed
-
- Braverman NE, D'Agostino MD, Maclean GE. Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives. Developmental disabilities research reviews. 2013;17:187–196. - PubMed
-
- Braverman N, Steel G, Obie C, Moser A, Moser H, Gould SJ, Valle D. Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata. Nature genetics. 1997;15:369–376. - PubMed
-
- Ebberink MS, Mooijer PA, Gootjes J, Koster J, Wanders RJ, Waterham HR. Genetic classification and mutational spectrum of more than 600 patients with a Zellweger syndrome spectrum disorder. Human mutation. 2011;32:59–69. - PubMed
-
- Ebberink MS, Koster J, Visser G, Spronsen F, Stolte-Dijkstra I, Smit GP, Fock JM, Kemp S, Wanders RJ, Waterham HR. A novel defect of peroxisome division due to a homozygous non-sense mutation in the PEX11beta gene. Journal of medical genetics. 2012;49:307–313. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases