

Evaluation of Novel Fetal Hemoglobin Inducer Drugs in Treatment of β-Hemoglobinopathy Disorders
- PMID: 24505535
- PMCID: PMC3913144
Evaluation of Novel Fetal Hemoglobin Inducer Drugs in Treatment of β-Hemoglobinopathy Disorders
Abstract
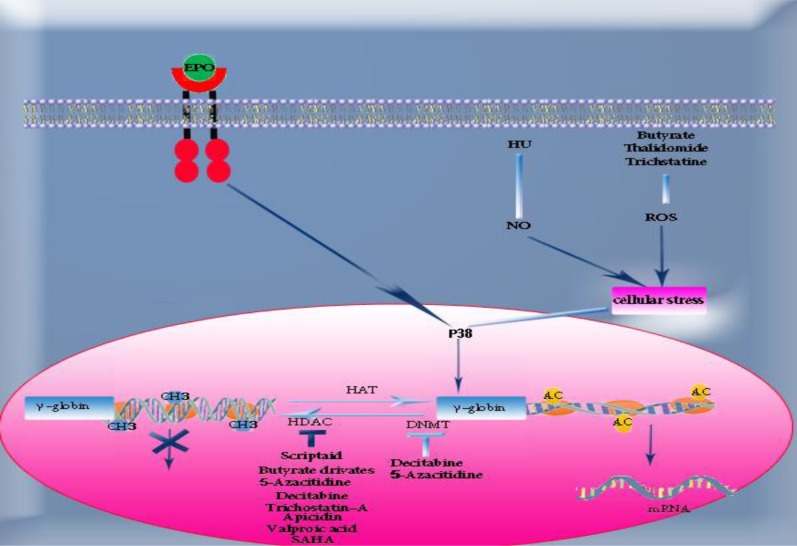
Objective: The use of fetal hemoglobin (HbF) inducer drugs is considered as a novel approach in treatment of β-hemoglobinopathies, especially β- thalassemia and sickle cell disease. HbF inducers including hydroxyurea, histone deacetylase (HDAC) inhibitor agents such as sodium butyrate, azacitidine, decitabine and new immunomodulator drugs like pomalidomide, lenalidomide and thalidomide can reduce α-globin chain production in erythroid progenitors and improve α: β chain imbalance, the most crucial complication of β-thalassemia.
Materials and methods: In this article, we reviewed more than 40 articles published from 1979 to 2012 in the field of fetal hemoglobin augmentation.
Results: Recent studies suggest the synergistic effect of drug combinations in efficient induction of fetal hemoglobin and gene over-expression.
Conclusion: It seems that drugs which act with different molecular and epigenetic mechanisms have proper synergistic effects in fetal hemoglobin induction and gene over-expression.
Keywords: Fetal hemoglobin; Histone deacetylase; β-Hemoglobinopathies.
Figures
Similar articles
-
Pomalidomide and lenalidomide regulate erythropoiesis and fetal hemoglobin production in human CD34+ cells.J Clin Invest. 2008 Jan;118(1):248-58. doi: 10.1172/JCI32322. J Clin Invest. 2008. PMID: 18064299 Free PMC article.
-
Thalidomide is more efficient than sodium butyrate in enhancing GATA-1 and EKLF gene expression in erythroid progenitors derived from HSCs with β-globin gene mutation.Int J Hematol Oncol Stem Cell Res. 2016 Jan 1;10(1):37-41. Int J Hematol Oncol Stem Cell Res. 2016. PMID: 27047649 Free PMC article.
-
Thiourea derivatives induce fetal hemoglobin production in-vitro: A new class of potential therapeutic agents for β-thalassemia.Eur J Pharmacol. 2019 Jul 15;855:285-293. doi: 10.1016/j.ejphar.2019.05.027. Epub 2019 May 14. Eur J Pharmacol. 2019. PMID: 31100414
-
The role of fetal hemoglobin-enhancing agents in thalassemia.Semin Hematol. 2004 Oct;41(4 Suppl 6):17-22. doi: 10.1053/j.seminhematol.2004.08.004. Semin Hematol. 2004. PMID: 15534853 Review.
-
Hemoglobinopathies.Hematology Am Soc Hematol Educ Program. 2003:14-39. doi: 10.1182/asheducation-2003.1.14. Hematology Am Soc Hematol Educ Program. 2003. PMID: 14633775 Review.
Cited by
-
Hydroxyurea down-regulates BCL11A, KLF-1 and MYB through miRNA-mediated actions to induce γ-globin expression: implications for new therapeutic approaches of sickle cell disease.Clin Transl Med. 2016 Mar;5(1):15. doi: 10.1186/s40169-016-0092-7. Epub 2016 Apr 7. Clin Transl Med. 2016. PMID: 27056246 Free PMC article.
-
Gamma reactivation using the spongy effect of KLF1-binding site sequence: an approach in gene therapy for beta-thalassemia.Iran J Basic Med Sci. 2016 Oct;19(10):1063-1069. Iran J Basic Med Sci. 2016. PMID: 27872702 Free PMC article.
-
Epigenetic effects toward new insights as potential therapeutic target in B-thalassemia.J Genet Eng Biotechnol. 2021 Mar 31;19(1):51. doi: 10.1186/s43141-021-00138-x. J Genet Eng Biotechnol. 2021. PMID: 33788050 Free PMC article.
-
Evaluation of the combination therapy of hydroxyurea and thalidomide in β-thalassemia.Blood Adv. 2022 Dec 27;6(24):6162-6168. doi: 10.1182/bloodadvances.2022007031. Blood Adv. 2022. PMID: 35477175 Free PMC article.
-
Molecular genetics of β-thalassemia: A narrative review.Medicine (Baltimore). 2021 Nov 12;100(45):e27522. doi: 10.1097/MD.0000000000027522. Medicine (Baltimore). 2021. PMID: 34766559 Free PMC article. Review.
References
-
- Flint J, Harding RM, Boyce AJ, Clegg JB. 1 The population genetics of the haemoglobinopathies. Baillière's clinical haematology. 1998;11(1):1–51. - PubMed
-
- Atweh GF, DeSimone J, Saunthararajah Y, Fathallah H, Weinberg RS, Nagel RL. Hemoglobinopathies. Hematology / the Education Program of the American Society of Hematology American Society of Hematology Education Program. 2003:14–39. PubMed PMID: 14633775. - PubMed
-
- Giardine B, van Baal S, Kaimakis P, Riemer C, Miller W, Samara M, et al. HbVar database of human hemoglobin variants and thalassemia mutations: 2007 update. Human Mutation. 2007;28(2):206. - PubMed
-
- Hardison RC, Chui DH, Riemer CR, Miller W, Carver MF, Molchanova TP, et al. Access to a syllabus of human hemoglobin variants (1996) via the World Wide Web. Hemoglobin. 1998 Mar;22(2):113–27. PubMed PMID: 9576329. - PubMed
-
- Karimi M, Yarmohammadi H, Farjadian S, Zeinali S, Moghaddam Z, Cappellini MD, et al. β-Thalassemia intermedia from southern Iran: IVS-II-1 (G→ A) is the prevalent thalassemia intermedia allele. Hemoglobin. 2002;26(2):147–54. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical