

A statistical framework to guide sequencing choices in pedigrees
- PMID: 24507777
- PMCID: PMC3928665
- DOI: 10.1016/j.ajhg.2014.01.005
A statistical framework to guide sequencing choices in pedigrees
Abstract
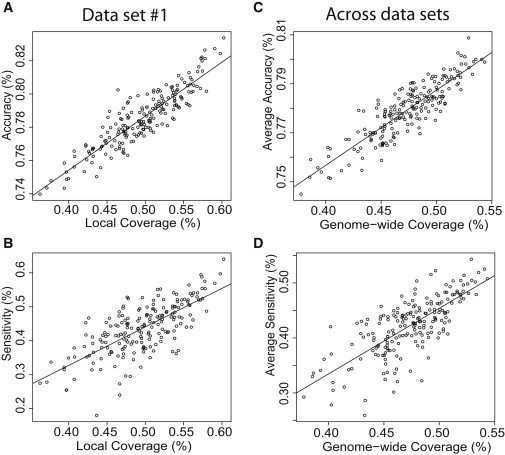
The use of large pedigrees is an effective design for identifying rare functional variants affecting heritable traits. Cost-effective studies using sequence data can be achieved via pedigree-based genotype imputation in which some subjects are sequenced and missing genotypes are inferred on the remaining subjects. Because of high cost, it is important to carefully prioritize subjects for sequencing. Here, we introduce a statistical framework that enables systematic comparison among subject-selection choices for sequencing. We introduce a metric "local coverage," which allows the use of inferred inheritance vectors to measure genotype-imputation ability specifically in a region of interest, such as one with prior evidence of linkage. In the absence of linkage information, we can instead use a "genome-wide coverage" metric computed with the pedigree structure. These metrics enable the development of a method that identifies efficient selection choices for sequencing. As implemented in GIGI-Pick, this method also flexibly allows initial manual selection of subjects and optimizes selections within the constraint that only some subjects might be available for sequencing. In the present study, we used simulations to compare GIGI-Pick with PRIMUS, ExomePicks, and common ad hoc methods of selecting subjects. In genotype imputation of both common and rare alleles, GIGI-Pick substantially outperformed all other methods considered and had the added advantage of incorporating prior linkage information. We also used a real pedigree to demonstrate the utility of our approach in identifying causal mutations. Our work enables prioritization of subjects for sequencing to facilitate dissection of the genetic basis of heritable traits.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures

Similar articles
-
GIGI: an approach to effective imputation of dense genotypes on large pedigrees.Am J Hum Genet. 2013 Apr 4;92(4):504-16. doi: 10.1016/j.ajhg.2013.02.011. Am J Hum Genet. 2013. PMID: 23561844 Free PMC article.
-
Power of family-based association designs to detect rare variants in large pedigrees using imputed genotypes.Genet Epidemiol. 2014 Jan;38(1):1-9. doi: 10.1002/gepi.21776. Epub 2013 Nov 15. Genet Epidemiol. 2014. PMID: 24243664 Free PMC article.
-
Combining family- and population-based imputation data for association analysis of rare and common variants in large pedigrees.Genet Epidemiol. 2014 Nov;38(7):579-90. doi: 10.1002/gepi.21844. Epub 2014 Aug 1. Genet Epidemiol. 2014. PMID: 25132070 Free PMC article.
-
Linkage analysis with sequential imputation.Genet Epidemiol. 2003 Jul;25(1):25-35. doi: 10.1002/gepi.10249. Genet Epidemiol. 2003. PMID: 12813724 Review.
-
Metropolis sampling in pedigree analysis.Stat Methods Med Res. 1993;2(3):263-82. doi: 10.1177/096228029300200305. Stat Methods Med Res. 1993. PMID: 8261261 Review.
Cited by
-
Identity-by-descent estimation with population- and pedigree-based imputation in admixed family data.BMC Proc. 2016 Oct 18;10(Suppl 7):295-301. doi: 10.1186/s12919-016-0046-5. eCollection 2016. BMC Proc. 2016. PMID: 27980652 Free PMC article.
-
Lessons learned from whole exome sequencing in multiplex families affected by a complex genetic disorder, intracranial aneurysm.PLoS One. 2015 Mar 24;10(3):e0121104. doi: 10.1371/journal.pone.0121104. eCollection 2015. PLoS One. 2015. PMID: 25803036 Free PMC article.
-
Revisit Population-based and Family-based Genotype Imputation.Sci Rep. 2019 Feb 12;9(1):1800. doi: 10.1038/s41598-018-38469-4. Sci Rep. 2019. PMID: 30755687 Free PMC article.
-
Whole-genome characterization in pedigreed non-human primates using genotyping-by-sequencing (GBS) and imputation.BMC Genomics. 2016 Aug 24;17(1):676. doi: 10.1186/s12864-016-2966-x. BMC Genomics. 2016. PMID: 27558348 Free PMC article.
-
Inferring Transmission Histories of Rare Alleles in Population-Scale Genealogies.Am J Hum Genet. 2018 Dec 6;103(6):893-906. doi: 10.1016/j.ajhg.2018.10.017. Am J Hum Genet. 2018. PMID: 30526866 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
