

Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants
- PMID: 24508459
- PMCID: PMC4361392
- DOI: 10.1016/j.celrep.2014.01.013
Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants
Abstract
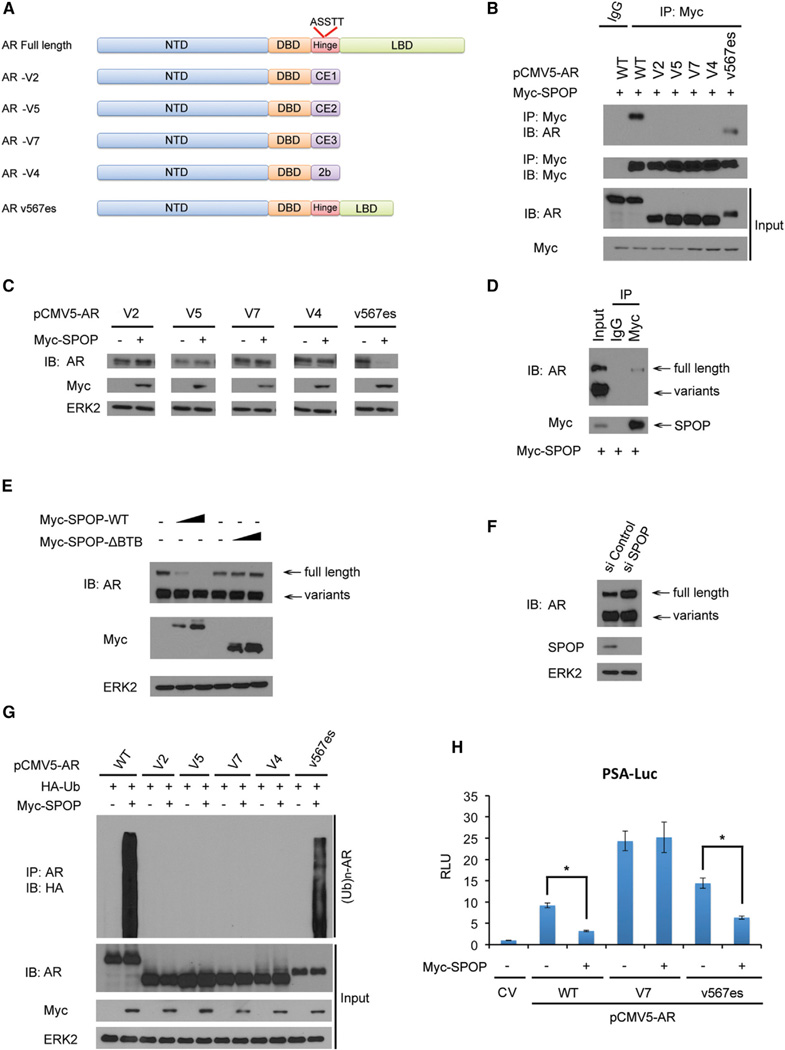
The SPOP E3 ubiquitin ligase gene is frequently mutated in human prostate cancers. Here, we demonstrate that SPOP recognizes a Ser/Thr-rich degron in the hinge domain of androgen receptor (AR) and induces degradation of full-length AR and inhibition of AR-mediated gene transcription and prostate cancer cell growth. AR splicing variants, most of which lack the hinge domain, escape SPOP-mediated degradation. Prostate-cancer-associated mutants of SPOP cannot bind to and promote AR destruction. Furthermore, androgens antagonize SPOP-mediated degradation of AR, whereas antiandrogens promote this process. This study identifies AR as a bona fide substrate of SPOP and elucidates a role of SPOP mutations in prostate cancer, thus implying the importance of this pathway in resistance to antiandrogen therapy of prostate cancer.
Copyright © 2014 The Authors. Published by Elsevier Inc. All rights reserved.
Figures






Comment in
-
Speckle-type POZ protein mutations interrupt tumor suppressor function of speckle-type POZ protein in prostate cancer by affecting androgen receptor degradation.Asian J Androl. 2014 Sep-Oct;16(5):659-60. doi: 10.4103/1008-682X.133323. Asian J Androl. 2014. PMID: 24969063 Free PMC article.
-
Two paths for stabilization of ERG in prostate carcinogenesis: TMPRSS2-ERG fusions and speckle-type pox virus and zinc finger protein mutations.Asian J Androl. 2016 Jul-Aug;18(4):594-5. doi: 10.4103/1008-682X.168793. Asian J Androl. 2016. PMID: 26763545 Free PMC article.
References
-
- Cai C, Chen S, Ng P, Bubley GJ, Nelson PS, Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–6513. - PMC - PubMed
-
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 2004;10:33–39. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
