

The deleterious mutation load is insensitive to recent population history
- PMID: 24509481
- PMCID: PMC3953611
- DOI: 10.1038/ng.2896
The deleterious mutation load is insensitive to recent population history
Abstract
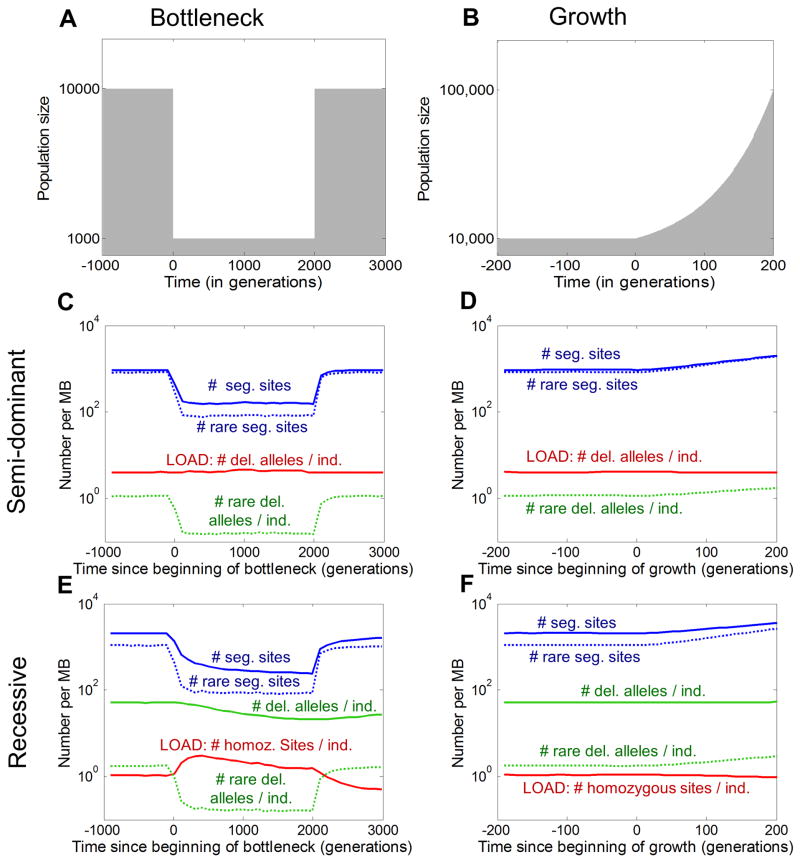
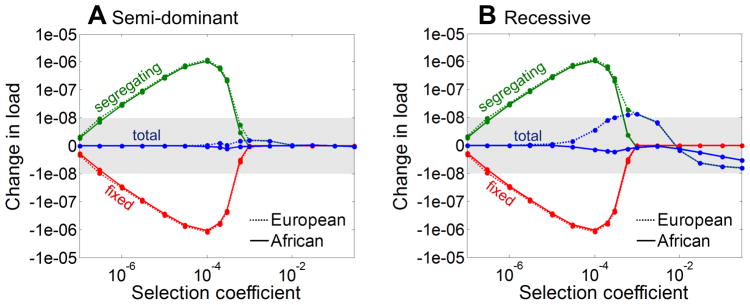
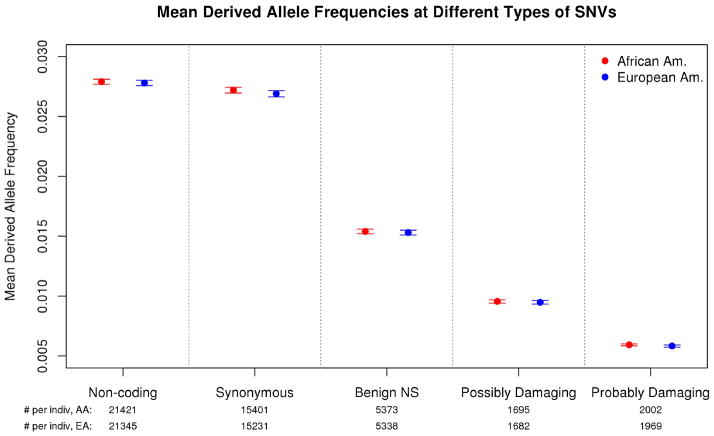
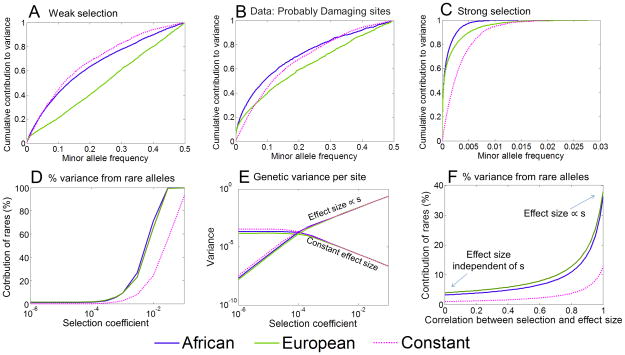
Human populations have undergone major changes in population size in the past 100,000 years, including recent rapid growth. How these demographic events have affected the burden of deleterious mutations in individuals and the frequencies of disease mutations in populations remains unclear. We use population genetic models to show that recent human demography has probably had little impact on the average burden of deleterious mutations. This prediction is supported by two exome sequence data sets showing that individuals of west African and European ancestry carry very similar burdens of damaging mutations. We further show that for many diseases, rare alleles are unlikely to contribute a large fraction of the heritable variation, and therefore the impact of recent growth is likely to be modest. However, for those diseases that have a direct impact on fitness, strongly deleterious rare mutations probably do have an important role, and recent growth will have increased their impact.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
