

Molecular mechanisms of varicella zoster virus pathogenesis
- PMID: 24509782
- PMCID: PMC4066823
- DOI: 10.1038/nrmicro3215
Molecular mechanisms of varicella zoster virus pathogenesis
Abstract
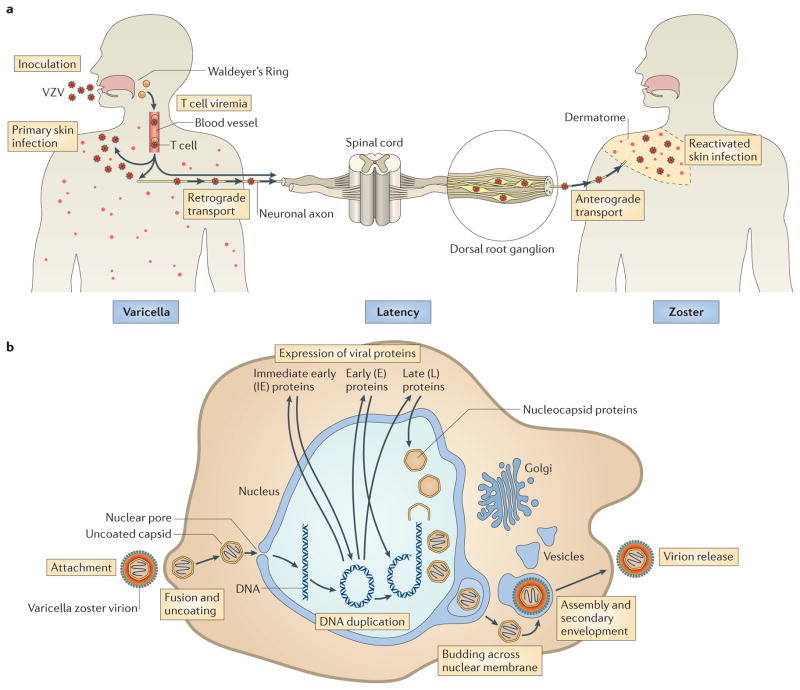
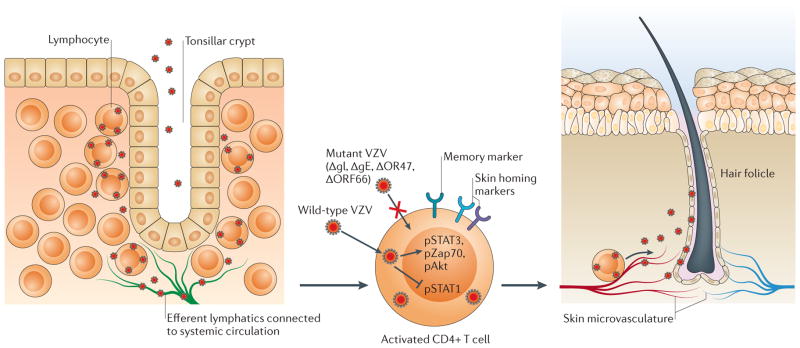
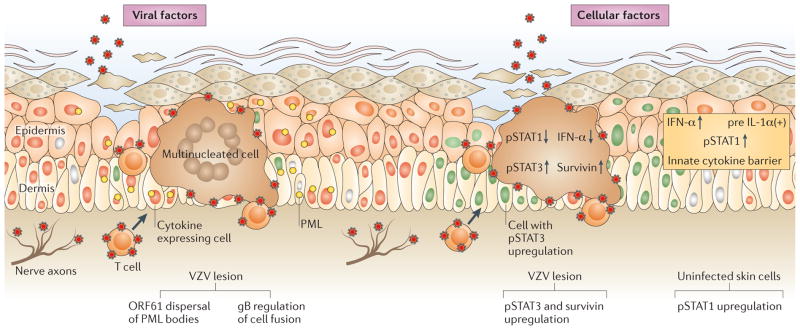
Varicella zoster virus (VZV) is the causative agent of varicella (chickenpox) and zoster (shingles). Investigating VZV pathogenesis is challenging as VZV is a human-specific virus and infection does not occur, or is highly restricted, in other species. However, the use of human tissue xenografts in mice with severe combined immunodeficiency (SCID) enables the analysis of VZV infection in differentiated human cells in their typical tissue microenvironment. Xenografts of human skin, dorsal root ganglia or foetal thymus that contains T cells can be infected with mutant viruses or in the presence of inhibitors of viral or cellular functions to assess the molecular mechanisms of VZV-host interactions. In this Review, we discuss how these models have improved our understanding of VZV pathogenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Arvin AM, Gilden D. In: Fields Virology. 6. Knipe D, Howley P, editors. Lippincott Williams & Wilkins; 2013. pp. 2015–2057.
-
- Ku CC, et al. Varicella-zoster virus transfer to skin by T cells and modulation of viral replication by epidermal cell interferon-α. J Exp Med. 2004;200:917–925. This paper shows the role of T cells in the transport of VZV and the control of replication by the potent innate response of skin cells. - PMC - PubMed
-
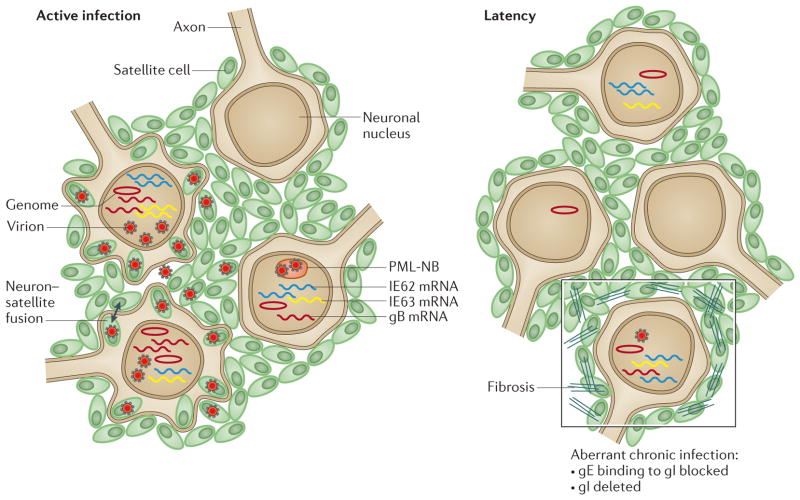
- Gilden DH, et al. Varicella-zoster virus DNA in human sensory ganglia. Nature. 1983;306:478–480. This study provides the first evidence of VZV latency in neurons, using in situ hybridization to detect VZV genomes. - PubMed
-
- Takahashi M. Clinical overview of varicella vaccine: development and early studies. Pediatrics. 1986;78:736–741. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
