

Cofilin drives cell-invasive and metastatic responses to TGF-β in prostate cancer
- PMID: 24509905
- PMCID: PMC4488067
- DOI: 10.1158/0008-5472.CAN-13-3058
Cofilin drives cell-invasive and metastatic responses to TGF-β in prostate cancer
Erratum in
-
Correction: Cofilin Drives Cell-Invasive and Metastatic Responses to TGF-β in Prostate Cancer.Cancer Res. 2018 Sep 1;78(17):5180. doi: 10.1158/0008-5472.CAN-18-1845. Cancer Res. 2018. PMID: 30181304 No abstract available.
Abstract
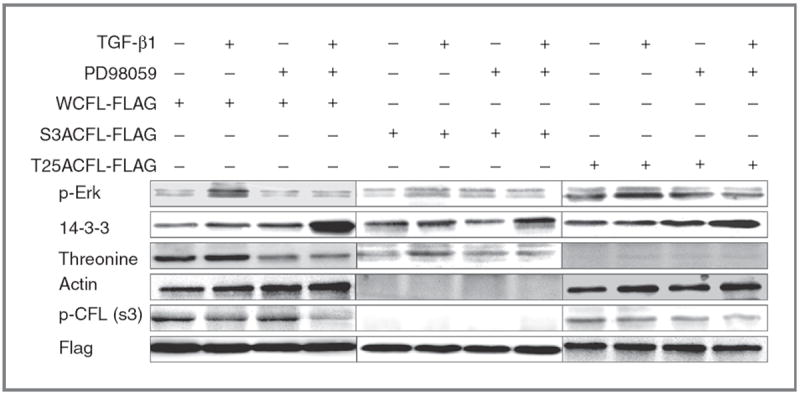
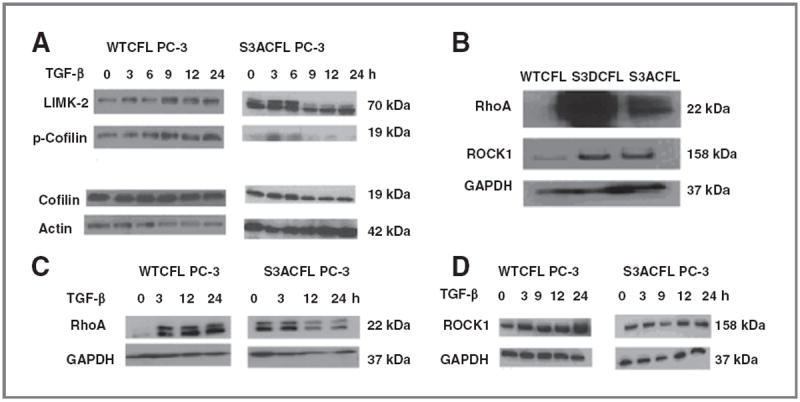
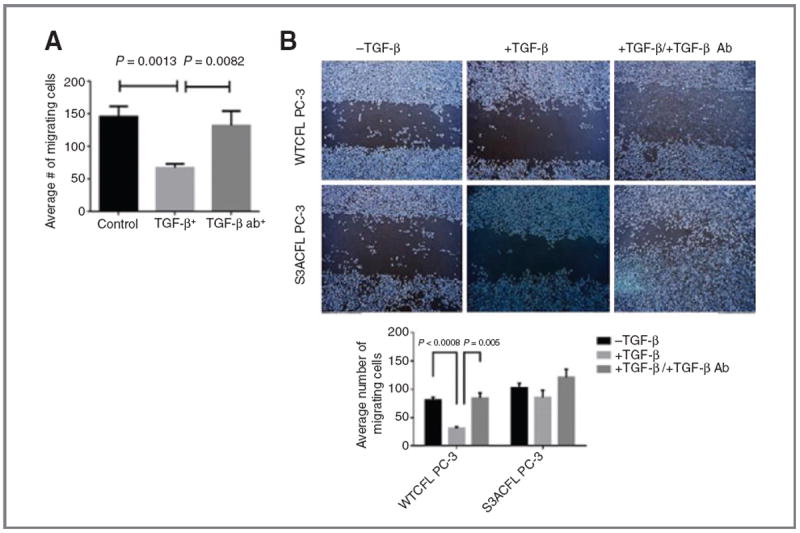
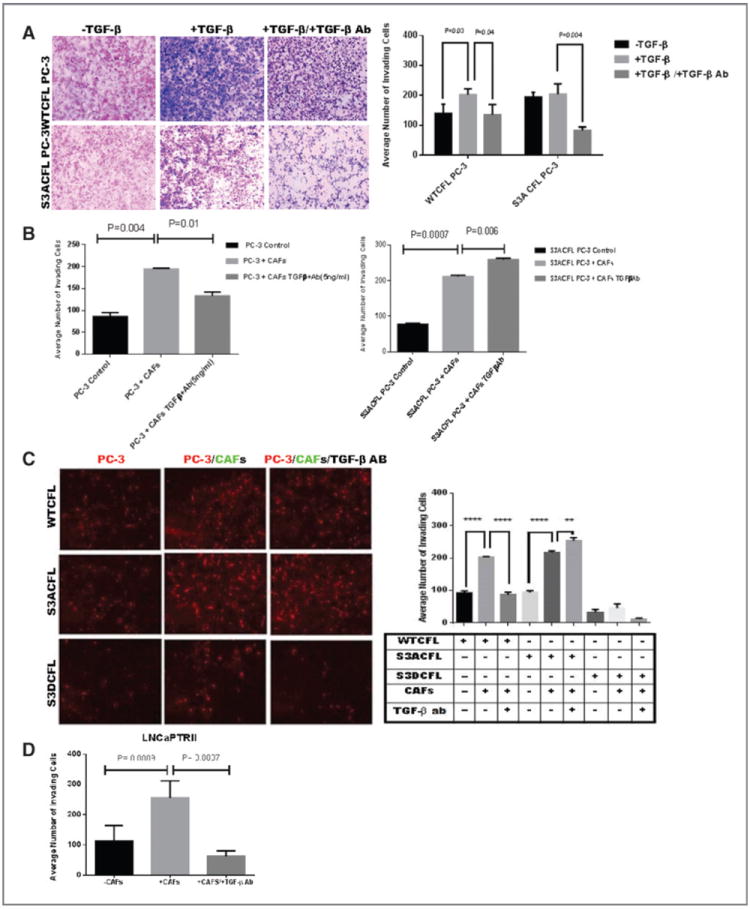
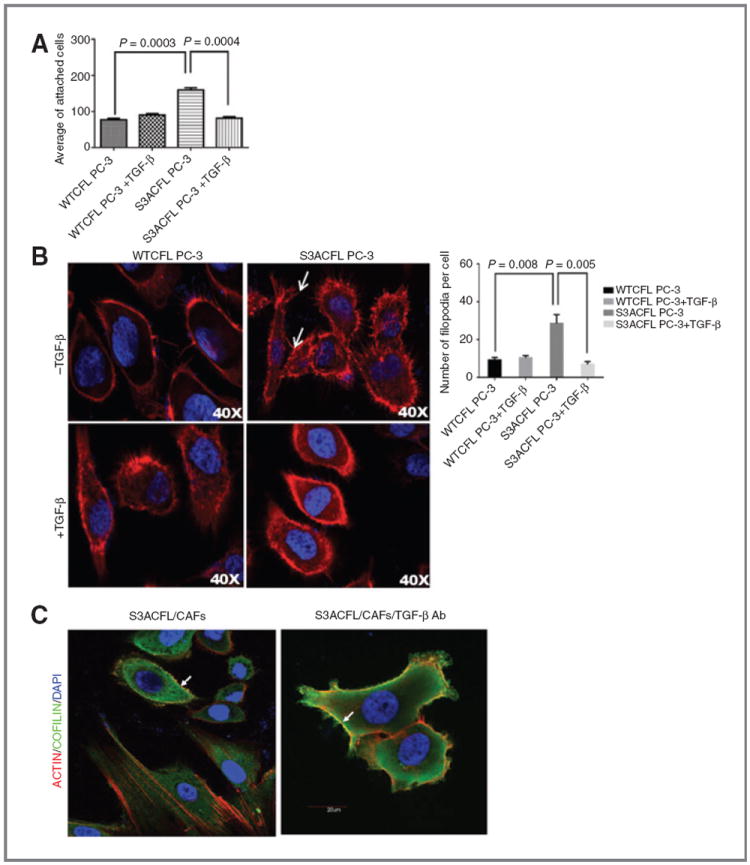
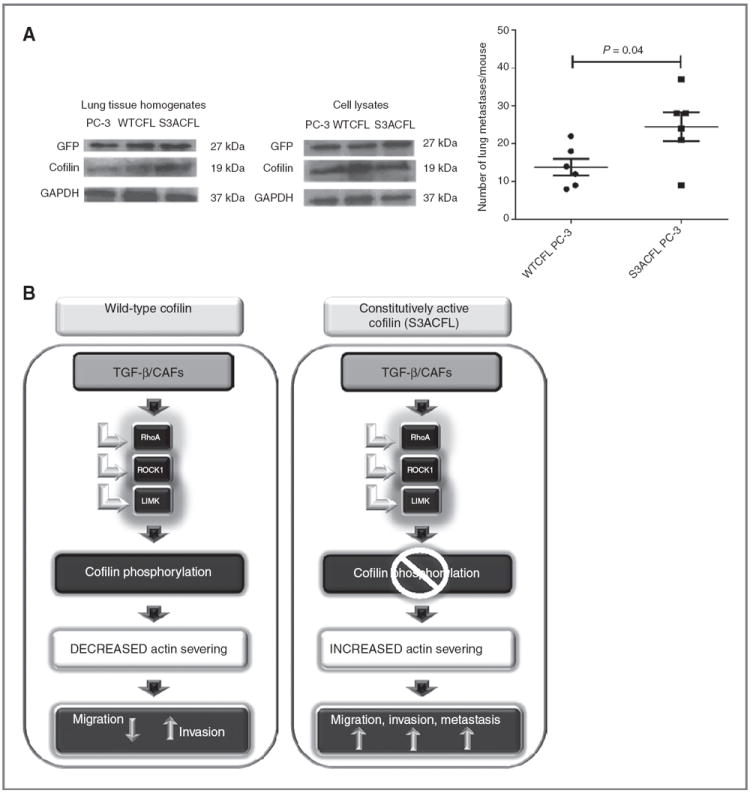
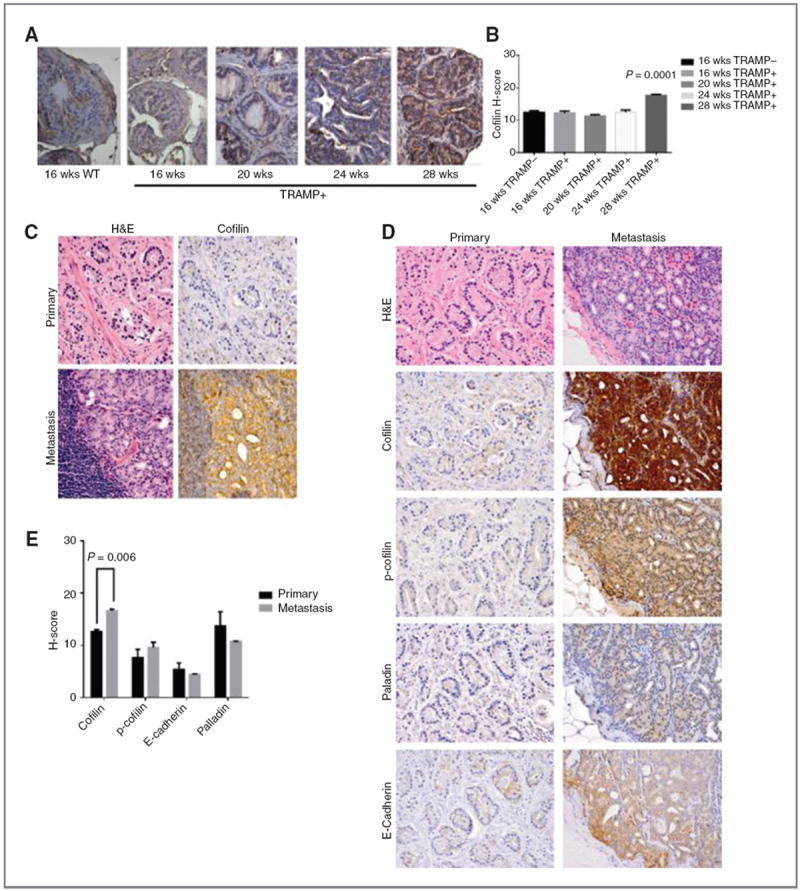
Cofilin (CFL) is an F-actin-severing protein required for the cytoskeleton reorganization and filopodia formation, which drives cell migration. CFL binding and severing of F-actin is controlled by Ser3 phosphorylation, but the contributions of this step to cell migration during invasion and metastasis of cancer cells are unclear. In this study, we addressed the question in prostate cancer cells, including the response to TGF-β, a critical regulator of migration. In cells expressing wild-type CFL, TGF-β treatment increased LIMK-2 activity and cofilin phosphorylation, decreasing filopodia formation. Conversely, constitutively active CFL (SerAla) promoted filipodia formation and cell migration mediated by TGF-β. Notably, in cocultures of prostate cancer epithelial cells and cancer-associated fibroblasts, active CFL promoted invasive migration in response to TGF-β in the microenvironment. Further, constitutively active CFL elevated the metastatic ability of prostate cancer cells in vivo. We found that levels of active CFL correlated with metastasis in a mouse model of prostate tumor and that in human prostate cancer, CFL expression was increased significantly in metastatic tumors. Our findings show that the actin-severing protein CFL coordinates responses to TGF-β that are needed for invasive cancer migration and metastasis.
©2014 AACR.
Conflict of interest statement
No potential conflicts of interest were disclosed.
Figures
References
-
- Catalona WJ. Management of cancer of the prostate. N Engl J Med. 1994;331:996–1004. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
