

Genome-scale sequencing to identify genes involved in Mendelian disorders
- PMID: 24510651
- PMCID: PMC3959778
- DOI: 10.1002/0471142905.hg0613s79
Genome-scale sequencing to identify genes involved in Mendelian disorders
Abstract
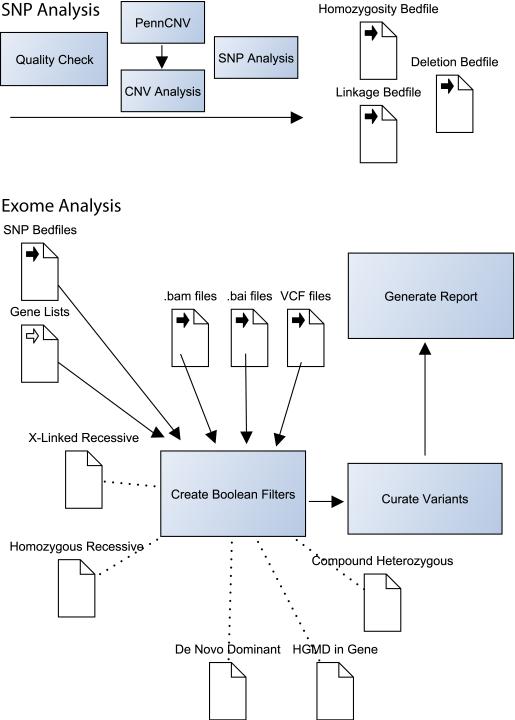
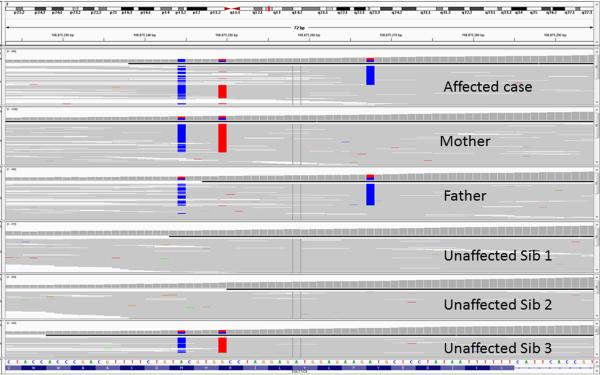
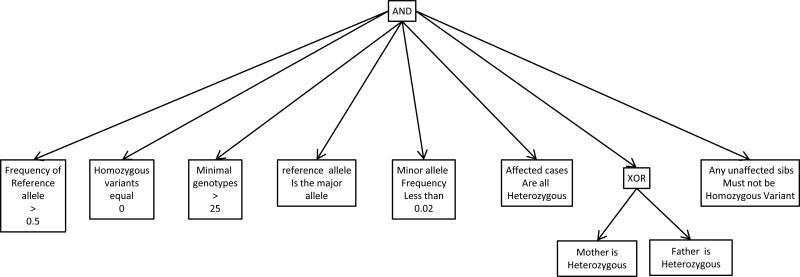
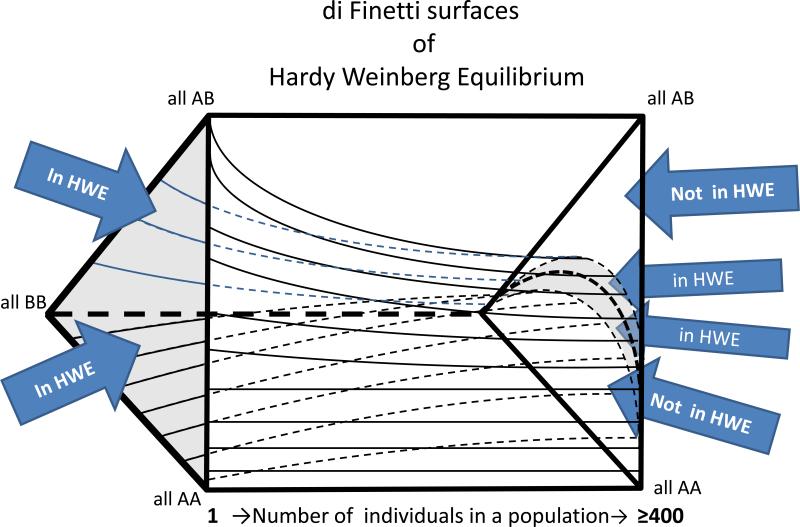
The analysis of genome-scale sequence data can be defined as the interrogation of a complete set of genetic instructions in a search for individual loci that produce or contribute to a pathological state. Bioinformatic analysis of sequence data requires sufficient discriminant power to find this needle in a haystack. Current approaches make choices about selectivity and specificity thresholds, and the quality, quantity, and completeness of the data in these analyses. There are many software tools available for individual, analytic component-tasks, including commercial and open-source options. Three major types of techniques have been included in most published exome projects to date: frequency/population genetic analysis, inheritance state consistency, and predictions of deleteriousness. The required infrastructure and use of each technique during analysis of genomic sequence data for clinical and research applications are discussed. Future developments will alter the strategies and sequence of using these tools and are also discussed.
Keywords: Mendelian inheritance; bioinformatics; clinical sequencing; exome; next generation sequencing.
Copyright © 2013 John Wiley & Sons, Inc.
Figures
References
-
- Anonymous . Online Mendelian Inheritance in Man, OMIM (TM) McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; National Center for Biotechnology Information, National Library of Medicine; Baltimore, MD: Bethesda, MD:
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
