

Homozygosity mapping reveals new nonsense mutation in the FAM161A gene causing autosomal recessive retinitis pigmentosa in a Palestinian family
- PMID: 24520187
- PMCID: PMC3919667
Homozygosity mapping reveals new nonsense mutation in the FAM161A gene causing autosomal recessive retinitis pigmentosa in a Palestinian family
Abstract
Purpose: Retinitis pigmentosa (RP) is a heterogenous group of inherited retinal degenerations caused by mutations in at least 45 genes. Recently, the FAM161A gene was identified as the causative gene for RP28, an autosomal recessive form of RP.
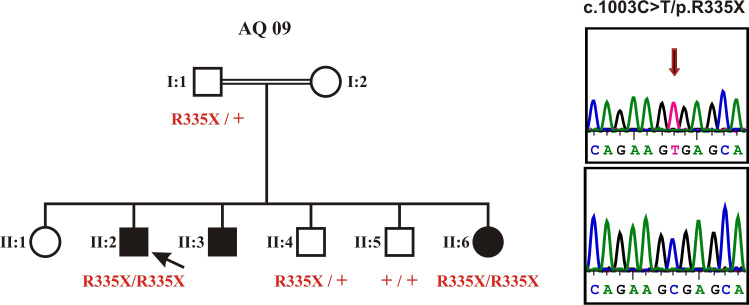
Methods: We performed a clinical and molecular genetic study of a consanguineous Palestinian family with two three siblings affected with retinitis pigmentosa. DNA samples were collected from the index patient, his father, his affected sister, and two non-affected brothers. DNA sample from the index was subjected to high resolution genome-wide SNP array. Assuming identity-by-descent in this consanguineous family we applied homozygosity mapping to identify disease causing genes.
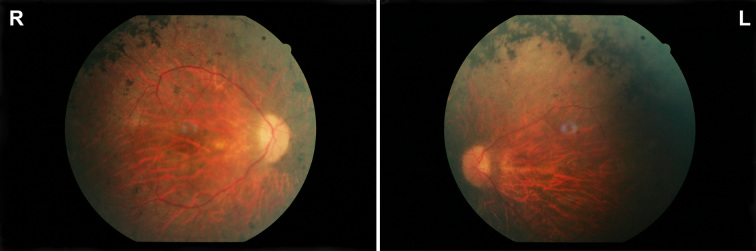
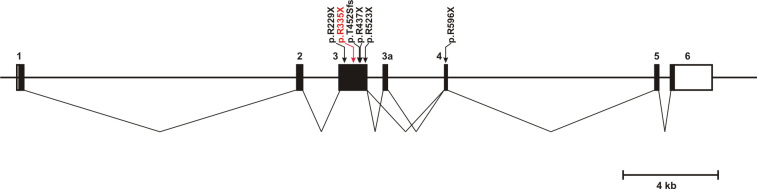
Results: The index patient reported night blindness since the age of 20 years, followed by moderate disease progression with decrease of peripheral vision, the development of photophobia and later on reduced central vision. At the age of 40 his visual acuity was counting fingers (CF) for both eyes, color discrimination was not possible and his visual fields were severely constricted. Funduscopic examination revealed a typical appearance of advanced RP with optic disc pallor, narrowed retinal vessels, bone-spicule like pigmentary changes in the mid-periphery and atrophic changes in the macula. His younger affected brother (37 years) was reported with overall milder symptoms, while the youngest sister (21 years) reported problems only with night vision. Applying high-density SNP arrays we identified several homozygous genomic regions one of which included the recently identified FAM161A gene mutated in RP28-linked autosomal recessive RP. Sequencing analysis revealed the presence of a novel homozygous nonsense mutation, c.1003C>T/p.R335X in the index patient and the affected sister.
Conclusion: We identified an RP28-linked RP family in the Palestinian population caused by a novel nonsense mutation in FAM161A. RP in this family shows a typical disease onset with moderate to rapid progression into severe visual impairment including central vision in the index and overall milder symptoms in the younger brother and sister.
Figures
Similar articles
-
Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa.Am J Hum Genet. 2010 Sep 10;87(3):382-91. doi: 10.1016/j.ajhg.2010.07.022. Epub 2010 Aug 12. Am J Hum Genet. 2010. PMID: 20705279 Free PMC article.
-
Whole-exome sequencing reveals a novel frameshift mutation in the FAM161A gene causing autosomal recessive retinitis pigmentosa in the Indian population.J Hum Genet. 2015 Oct;60(10):625-30. doi: 10.1038/jhg.2015.92. Epub 2015 Aug 6. J Hum Genet. 2015. PMID: 26246154
-
Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa.Am J Hum Genet. 2010 Sep 10;87(3):376-81. doi: 10.1016/j.ajhg.2010.07.018. Epub 2010 Aug 12. Am J Hum Genet. 2010. PMID: 20705278 Free PMC article.
-
FAM161A, a novel centrosomal-ciliary protein implicated in autosomal recessive retinitis pigmentosa.Adv Exp Med Biol. 2014;801:185-90. doi: 10.1007/978-1-4614-3209-8_24. Adv Exp Med Biol. 2014. PMID: 24664697 Review.
-
Optical Coherence Tomography (OCT) Diagnostic of Retinitis Pigmentosa - Case Study.Acta Inform Med. 2022 Dec;30(4):329-333. doi: 10.5455/aim.2022.30.329-333. Acta Inform Med. 2022. PMID: 36467319 Free PMC article. Review.
Cited by
-
Morphological and Functional Comparison of Mice Models for Retinitis Pigmentosa.Adv Exp Med Biol. 2023;1415:365-370. doi: 10.1007/978-3-031-27681-1_53. Adv Exp Med Biol. 2023. PMID: 37440058 Review.
-
Unique combination of clinical features in a large cohort of 100 patients with retinitis pigmentosa caused by FAM161A mutations.Sci Rep. 2020 Sep 16;10(1):15156. doi: 10.1038/s41598-020-72028-0. Sci Rep. 2020. PMID: 32938956 Free PMC article.
-
The genetics of rod-cone dystrophy in Arab countries: a systematic review.Eur J Hum Genet. 2021 Jun;29(6):897-910. doi: 10.1038/s41431-020-00754-0. Epub 2020 Nov 13. Eur J Hum Genet. 2021. PMID: 33188265 Free PMC article.
-
Insight into Ocular Genetic Research: Trends in Oman.Oman Med J. 2015 May;30(3):149-50. doi: 10.5001/omj.2015.33. Oman Med J. 2015. PMID: 26171118 Free PMC article. No abstract available.
-
Swept-source optical coherence tomography changes and visual acuity among Palestinian retinitis Pigmentosa patients: a cross-sectional study.BMC Ophthalmol. 2021 Jul 29;21(1):289. doi: 10.1186/s12886-021-02047-6. BMC Ophthalmol. 2021. PMID: 34320936 Free PMC article.
References
-
- Zobor D, Zrenner E. Retinitis pigmentosa - a review. Pathogenesis, guidelines for diagnostics and perspectives. Ophthalmologe. 2012;109:501–14. - PubMed
-
- Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29:335–75. - PubMed
-
- Janecke AR, Thompson DA, Utermann G, Becker C, Hübner CA, Schmid E, McHenry CL, Nair AR, Rüschendorf F, Heckenlively J, Wissinger B, Nürnberg P, Gal A. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet. 2004;36:850–4. - PubMed
-
- Zlotogora J. Genetic disorders among Palestinian Arabs: 1. Effects of consanguinity. Am J Med Genet. 1997;68:472–5. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
