

Non-muscle Mlck is required for β-catenin- and FoxO1-dependent downregulation of Cldn5 in IL-1β-mediated barrier dysfunction in brain endothelial cells
- PMID: 24522189
- PMCID: PMC4074294
- DOI: 10.1242/jcs.144550
Non-muscle Mlck is required for β-catenin- and FoxO1-dependent downregulation of Cldn5 in IL-1β-mediated barrier dysfunction in brain endothelial cells
Abstract
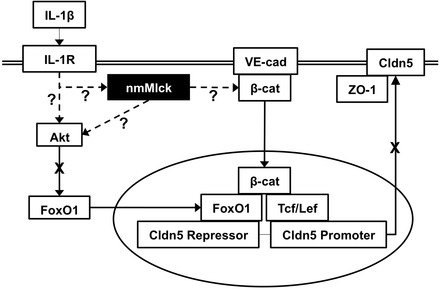
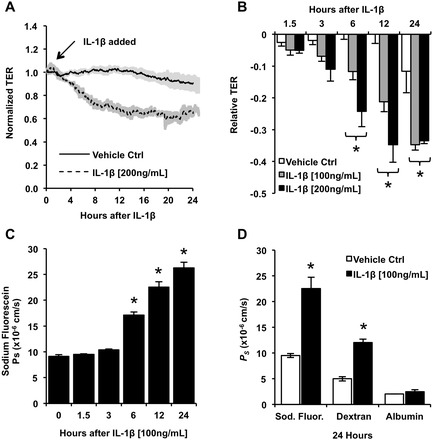
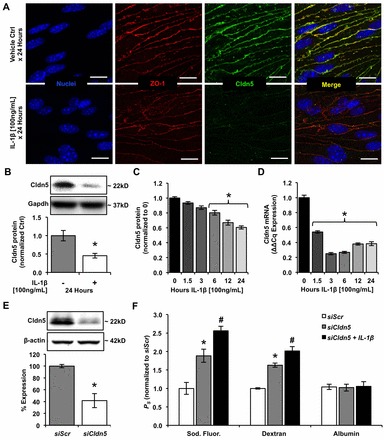
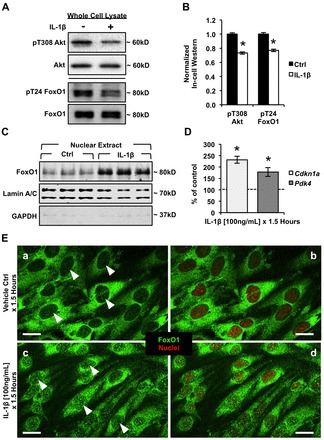
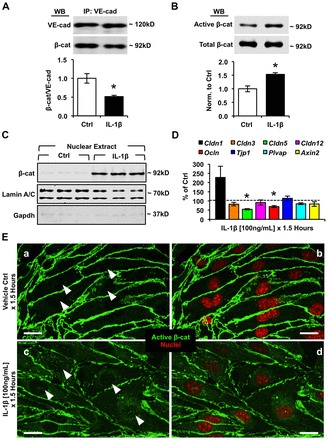
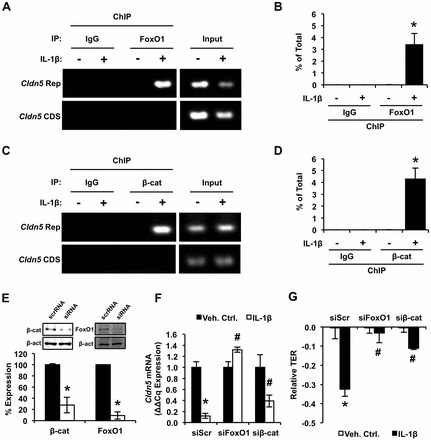
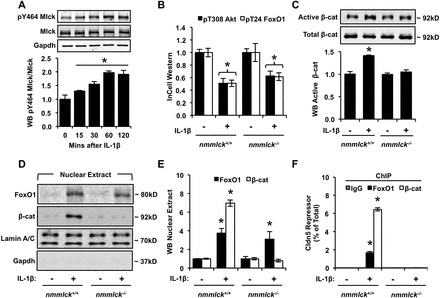
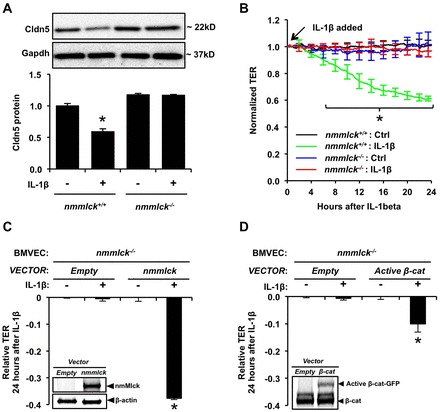
Aberrant elevation in the levels of the pro-inflammatory cytokine interleukin-1β (IL-1β) contributes to neuroinflammatory diseases. Blood-brain barrier (BBB) dysfunction is a hallmark phenotype of neuroinflammation. It is known that IL-1β directly induces BBB hyperpermeability but the mechanisms remain unclear. Claudin-5 (Cldn5) is a tight junction protein found at endothelial cell-cell contacts that are crucial for maintaining brain microvascular endothelial cell (BMVEC) integrity. Transcriptional regulation of Cldn5 has been attributed to the transcription factors β-catenin and forkhead box protein O1 (FoxO1), and the signaling molecules regulating their nuclear translocation. Non-muscle myosin light chain kinase (nmMlck, encoded by the Mylk gene) is a key regulator involved in endothelial hyperpermeability, and IL-1β has been shown to mediate nmMlck-dependent barrier dysfunction in epithelia. Considering these factors, we tested the hypothesis that nmMlck modulates IL-1β-mediated downregulation of Cldn5 in BMVECs in a manner that depends on transcriptional repression mediated by β-catenin and FoxO1. We found that treating BMVECs with IL-1β induced barrier dysfunction concomitantly with the nuclear translocation of β-catenin and FoxO1 and the repression of Cldn5. Most importantly, using primary BMVECs isolated from mice null for nmMlck, we identified that Cldn5 repression caused by β-catenin and FoxO1 in IL-1β-mediated barrier dysfunction was dependent on nmMlck.
Keywords: Blood–brain barrier; Claudin-5; FoxO1; IL-1β; Neuroinflammation; Non-muscle myosin light chain kinase; β-catenin.
Figures
Similar articles
-
AKT2 maintains brain endothelial claudin-5 expression and selective activation of IR/AKT2/FOXO1-signaling reverses barrier dysfunction.J Cereb Blood Flow Metab. 2020 Feb;40(2):374-391. doi: 10.1177/0271678X18817512. Epub 2018 Dec 21. J Cereb Blood Flow Metab. 2020. PMID: 30574832 Free PMC article.
-
Progression of experimental autoimmune encephalomyelitis in mice and neutrophil-mediated blood-brain barrier dysfunction requires non-muscle myosin light chain kinase.J Cereb Blood Flow Metab. 2025 Jun;45(6):1203-1220. doi: 10.1177/0271678X251318620. Epub 2025 Feb 7. J Cereb Blood Flow Metab. 2025. PMID: 39917847 Free PMC article.
-
Endothelial dysfunction and claudin 5 regulation during acrolein-induced lung injury.Am J Respir Cell Mol Biol. 2011 Apr;44(4):483-90. doi: 10.1165/rcmb.2009-0391OC. Epub 2010 Jun 4. Am J Respir Cell Mol Biol. 2011. PMID: 20525806 Free PMC article.
-
The region-selective regulation of endothelial claudin-5 expression and signaling in brain health and disorders.J Cell Physiol. 2021 Oct;236(10):7134-7143. doi: 10.1002/jcp.30357. Epub 2021 Mar 10. J Cell Physiol. 2021. PMID: 33694168 Review.
-
Myosin light chain kinase in microvascular endothelial barrier function.Cardiovasc Res. 2010 Jul 15;87(2):272-80. doi: 10.1093/cvr/cvq144. Epub 2010 May 17. Cardiovasc Res. 2010. PMID: 20479130 Free PMC article. Review.
Cited by
-
Therapeutic Target and Cell-signal Communication of Chlorpromazine and Promethazine in Attenuating Blood-Brain Barrier Disruption after Ischemic Stroke.Cell Transplant. 2019 Feb;28(2):145-156. doi: 10.1177/0963689718819443. Epub 2018 Dec 20. Cell Transplant. 2019. PMID: 30569751 Free PMC article.
-
The Regulation of Intestinal Mucosal Barrier by Myosin Light Chain Kinase/Rho Kinases.Int J Mol Sci. 2020 May 18;21(10):3550. doi: 10.3390/ijms21103550. Int J Mol Sci. 2020. PMID: 32443411 Free PMC article. Review.
-
Foxo1 deletion promotes the growth of new lymphatic valves.J Clin Invest. 2021 Jul 15;131(14):e142341. doi: 10.1172/JCI142341. J Clin Invest. 2021. PMID: 34263740 Free PMC article.
-
Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction.Front Cell Neurosci. 2021 Sep 13;15:661838. doi: 10.3389/fncel.2021.661838. eCollection 2021. Front Cell Neurosci. 2021. PMID: 34588955 Free PMC article. Review.
-
Brain barriers: Crosstalk between complex tight junctions and adherens junctions.J Cell Biol. 2015 May 25;209(4):493-506. doi: 10.1083/jcb.201412147. J Cell Biol. 2015. PMID: 26008742 Free PMC article. Review.
References
-
- Barbieri S. S., Weksler B. B. (2007). Tobacco smoke cooperates with interleukin-1beta to alter beta-catenin trafficking in vascular endothelium resulting in increased permeability and induction of cyclooxygenase-2 expression in vitro and in vivo. FASEB J. 21, 1831–1843 10.1096/fj.06-7557com - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
