

Pheochromocytoma and paraganglioma syndromes: genetics and management update
- PMID: 24523625
- PMCID: PMC3921052
- DOI: 10.3747/co.21.1579
Pheochromocytoma and paraganglioma syndromes: genetics and management update
Abstract
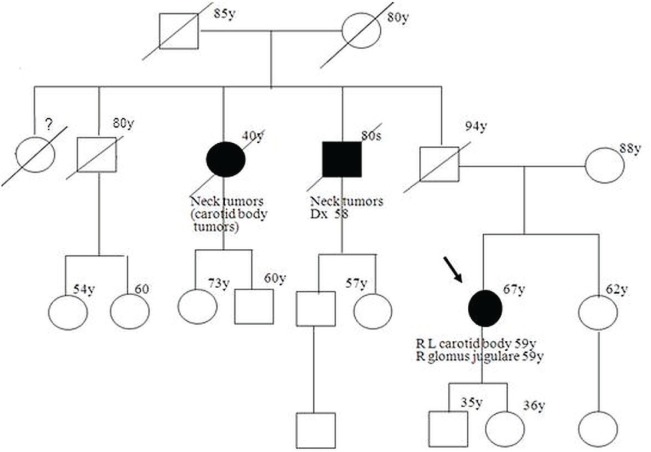
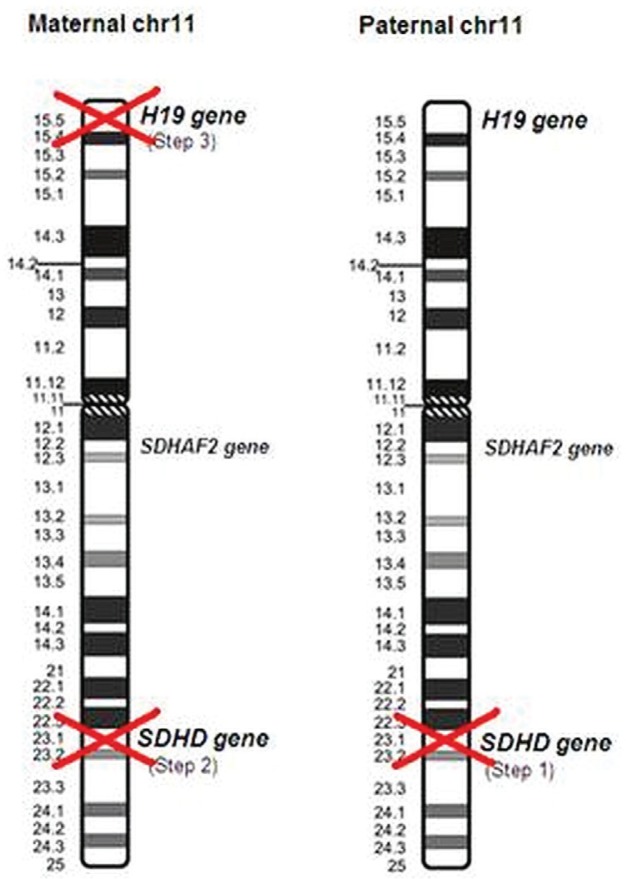
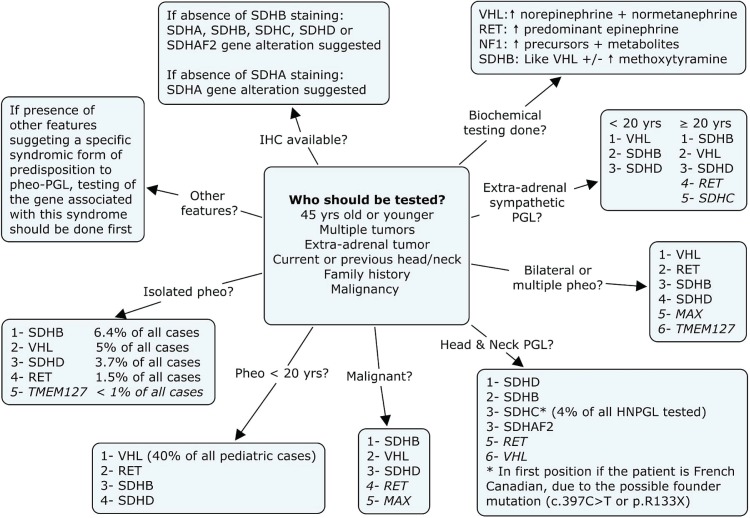
Pheochromocytomas (pheos) and paragangliomas (pgls) are rare tumours of the autonomic nervous system, originating from paraganglia, which are dispersed neuroendocrine organs characterized by catecholamine and peptide-producing cells derived from the neural crest. Medical textbooks have traditionally suggested that 10% of pheos are heritable. However, the frequency of heritable pheo has been underestimated. Three syndromic conditions-Von Hippel-Lindau (vhl), multiple endocrine neoplasia type 2 (men2), and neurofibromatosis type 1 (nf1)-and three genes-subunits of the succinate dehydrogenase (SDH) complex: SDHB, SDHC, and SDHD-are established causes of hereditary pheo-pgl. In the last few years, four new genes (SDHA, SDHAF2, MAX, and TMEM127) have been found to be associated with predisposition to these tumours. The present review, illustrated by three case reports, gives an update of the genetic basis of pheo-pgl and of the parent-of-origin effect implicated in the transmission of SDHD and SDHAF2. We discuss the referral criteria that should guide the decision to offer genetic testing to affected patients. We also specify the genes that are most likely implicated-based on particular features such as malignancy, bilateralism, or childhood-onset-to help geneticists efficiently order appropriate genetic tests. Finally, we review the screening recommendations for carriers of a pheo-pgl predisposition mutation.
Keywords: Pheochromocytoma; genetics; management; paraganglioma; parent-of-origin effect; predisposition; screening; testing.
Figures
References
-
- Dumas N, Edmont M, Grunenwald S, et al. Characterisation of genetics aspects of paragangliomas and pheochromocytomas in the Quebec French-Canadian population. Presented at the 12th International Congress of Human Genetics; Montreal, QC. October 11–15, 2011; [Available online at: http://www.ichg2011.org/cgi-bin/showdetail.pl?absno=10669; cited June 17, 2013]
-
- Kirmani S, Young WF. Hereditary paraganglioma–pheochromocytoma syndromes. In: Pagon RA, Bird TD, Dolan CR, et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993. [Available online at: http://www.ncbi.nlm.nih.gov/books/NBK1548; cited August 30, 2012] - PubMed
-
- DeLellis RA, Lloyd RV, Heitz PU, Eng C, editors. WHO Classification of Tumours Pathology and Genetics Tumours of Endocrine Organs. Lyon, France: IARC Press; 2004.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
