

Matrix revisited: mechanisms linking energy substrate metabolism to the function of the heart
- PMID: 24526677
- PMCID: PMC4410983
- DOI: 10.1161/CIRCRESAHA.114.301863
Matrix revisited: mechanisms linking energy substrate metabolism to the function of the heart
Abstract
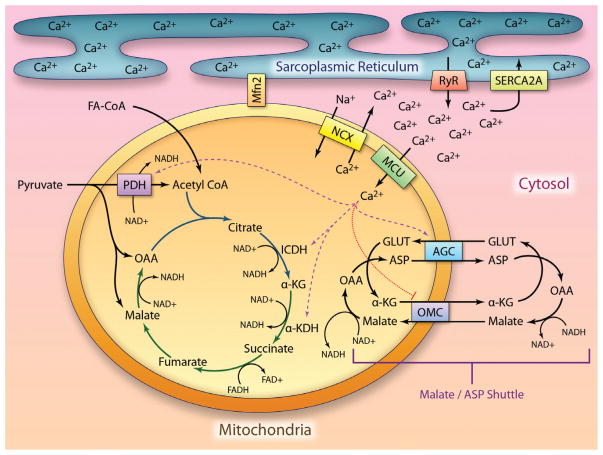
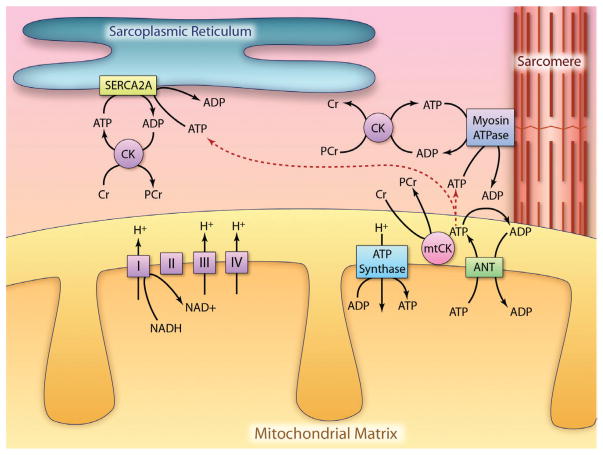
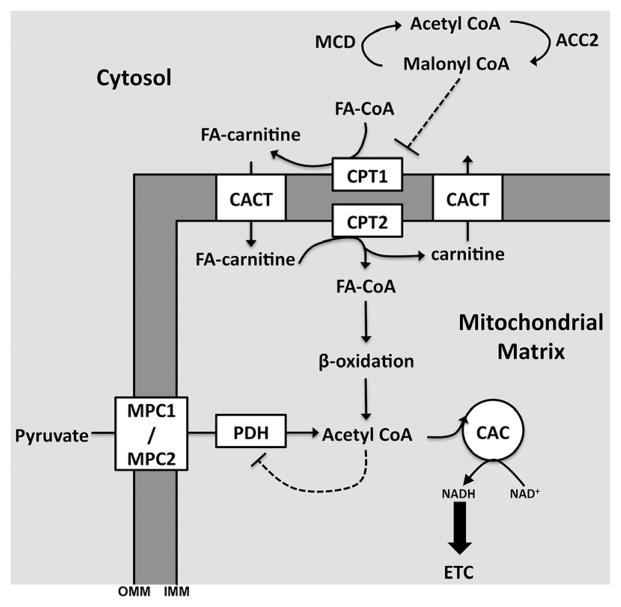
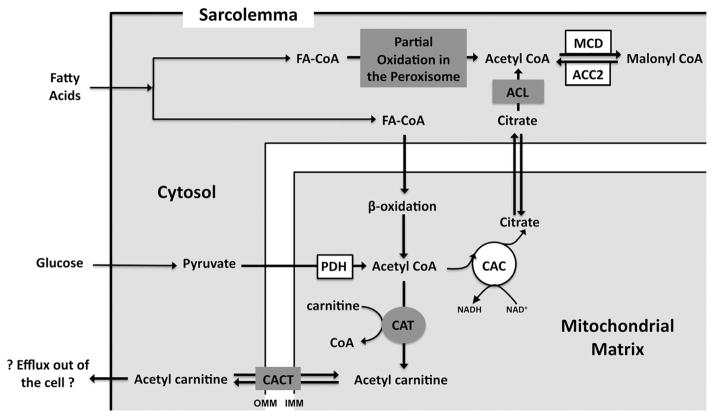
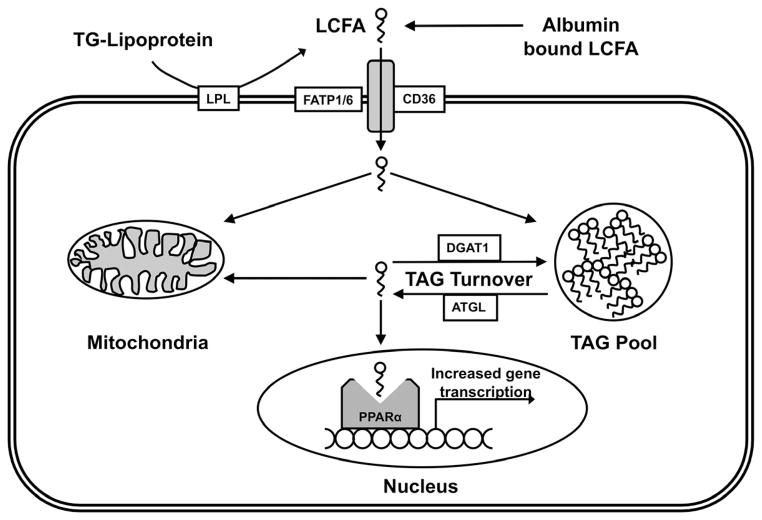
Metabolic signaling mechanisms are increasingly recognized to mediate the cellular response to alterations in workload demand, as a consequence of physiological and pathophysiological challenges. Thus, an understanding of the metabolic mechanisms coordinating activity in the cytosol with the energy-providing pathways in the mitochondrial matrix becomes critical for deepening our insights into the pathogenic changes that occur in the stressed cardiomyocyte. Processes that exchange both metabolic intermediates and cations between the cytosol and mitochondria enable transduction of dynamic changes in contractile state to the mitochondrial compartment of the cell. Disruption of such metabolic transduction pathways has severe consequences for the energetic support of contractile function in the heart and is implicated in the pathogenesis of heart failure. Deficiencies in metabolic reserve and impaired metabolic transduction in the cardiomyocyte can result from inherent deficiencies in metabolic phenotype or maladaptive changes in metabolic enzyme expression and regulation in the response to pathogenic stress. This review examines both current and emerging concepts of the functional linkage between the cytosol and the mitochondrial matrix with a specific focus on metabolic reserve and energetic efficiency. These principles of exchange and transport mechanisms across the mitochondrial membrane are reviewed for the failing heart from the perspectives of chronic pressure overload and diabetes mellitus.
Keywords: cytosol; diabetes mellitus; heart failure; metabolic pathways; metabolism; mitochondria; mobilization.
Figures
References
-
- Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic calcium. J Mol Cell Cardiol. 2002;34:1259–1271. - PubMed
-
- Mootha VK, Arai AE, Balaban RS. Maximum oxidative phosphorylation capacity of the mammalian heart. Am J Physiol. 1997;272:H769–H775. - PubMed
-
- Bache RJ, Zhang J, Murakami Y, Zhang Y, Cho YK, Merkle H, Gong G, From AH, Ugurbil K. Myocardial oxygenation at high workstates in hearts with left ventricular hypertrophy. Cardiovasc Res. 1999;42:616–626. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
