

Dynamics of Transforming Growth Factor Beta Signaling in Wound Healing and Scarring
- PMID: 24527343
- PMCID: PMC3857355
- DOI: 10.1089/wound.2013.0429
Dynamics of Transforming Growth Factor Beta Signaling in Wound Healing and Scarring
Abstract
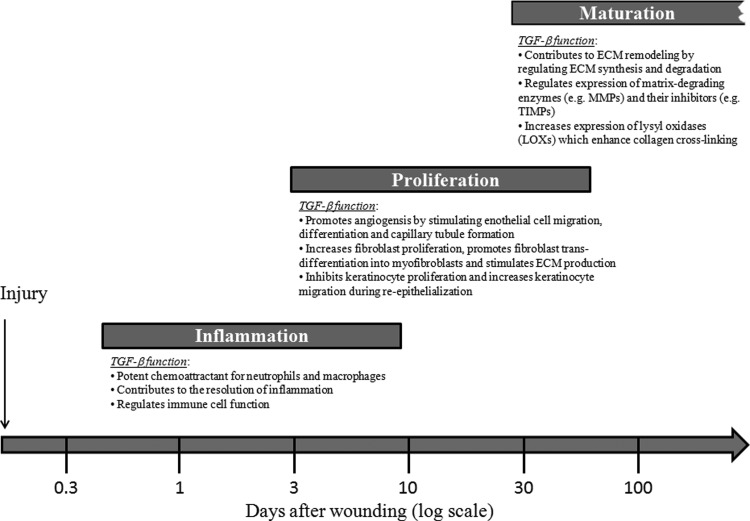
Significance: Wound healing is an intricate biological process in which the skin, or any other tissue, repairs itself after injury. Normal wound healing relies on the appropriate levels of cytokines and growth factors to ensure that cellular responses are mediated in a coordinated manner. Among the many growth factors studied in the context of wound healing, transforming growth factor beta (TGF-β) is thought to have the broadest spectrum of effects.
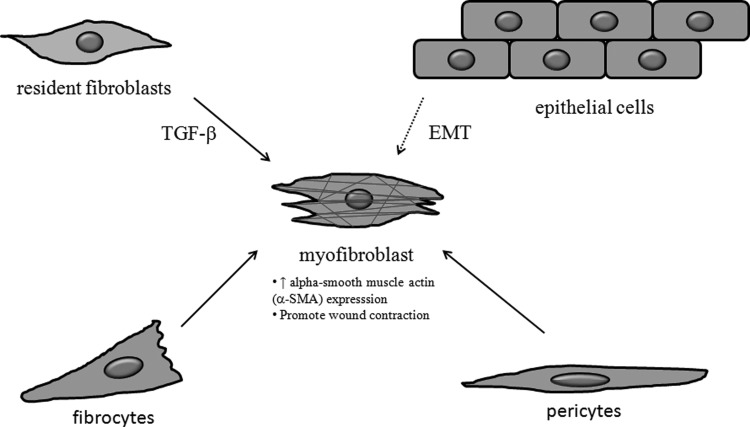
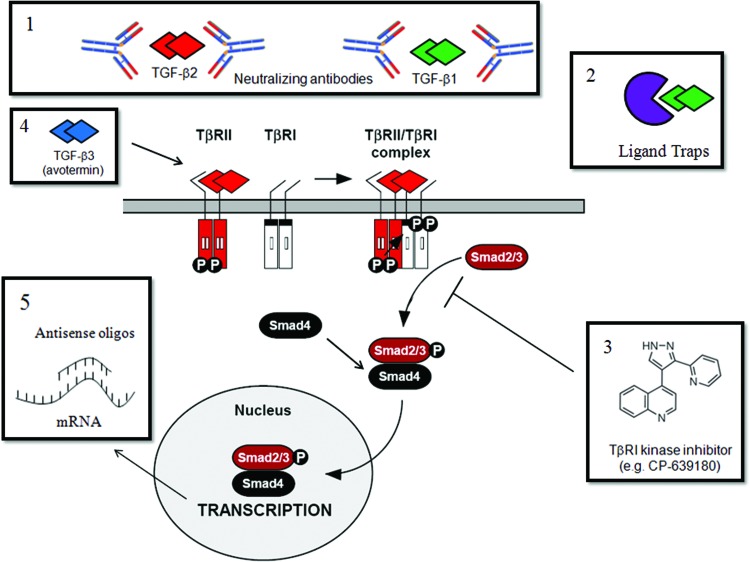
Recent advances: Many of the molecular mechanisms underlying the TGF-β/Smad signaling pathway have been elucidated, and the role of TGF-β in wound healing has been well characterized. Targeting the TGF-β signaling pathway using therapeutic agents to improve wound healing and/or reduce scarring has been successful in pre-clinical studies.
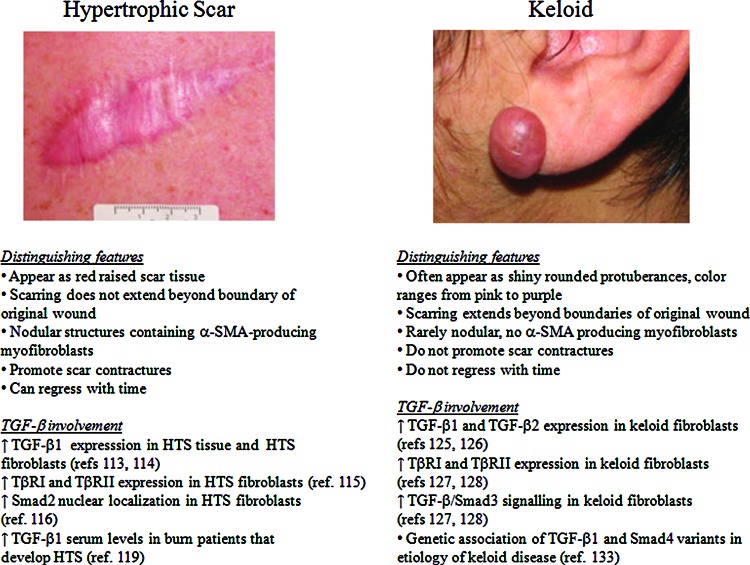
Critical issues: Although TGF-β isoforms (β1, β2, β3) signal through the same cell surface receptors, they display distinct functions during wound healing in vivo through mechanisms that have not been fully elucidated. The challenge of translating preclinical studies targeting the TGF-β signaling pathway to a clinical setting may require more extensive preclinical research using animal models that more closely mimic wound healing and scarring in humans, and taking into account the spatial, temporal, and cell-type-specific aspects of TGF-β isoform expression and function.
Future directions: Understanding the differences in TGF-β isoform signaling at the molecular level and identification of novel components of the TGF-β signaling pathway that critically regulate wound healing may lead to the discovery of potential therapeutic targets for treatment of impaired wound healing and pathological scarring.
Figures

References
-
- Wu MY. Hill CS. TGF-β superfamily signaling in embryonic development and homeostasis. Dev Cell. 2009;16:329. - PubMed
-
- Rider CC. Mulloy B. Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem J. 2010;429:1. - PubMed
-
- Schmierer B. Hill CS. TGF-β/Smad signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol. 2007;8:970. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
