

Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood
- PMID: 24530203
- PMCID: PMC3951944
- DOI: 10.1016/j.ajhg.2014.01.006
Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood
Abstract
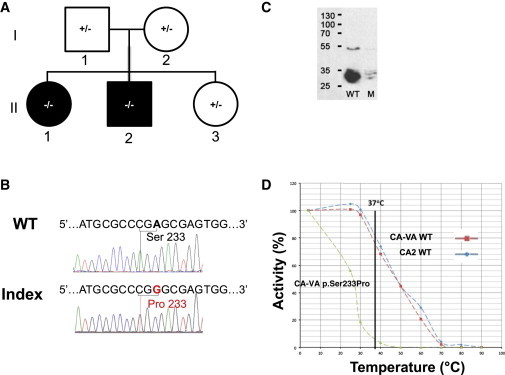
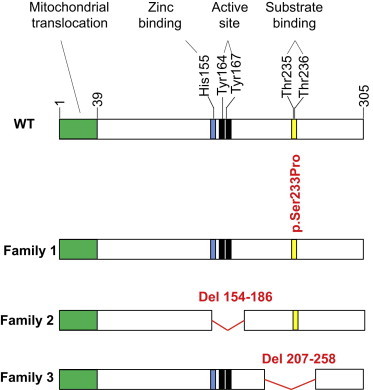
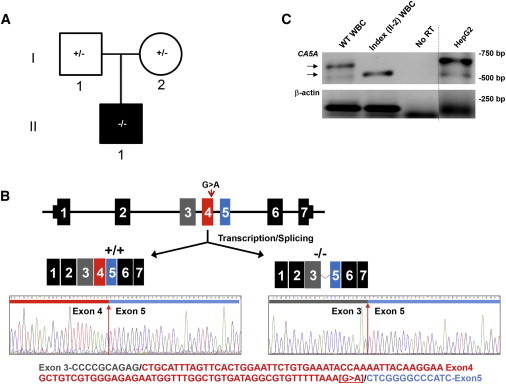
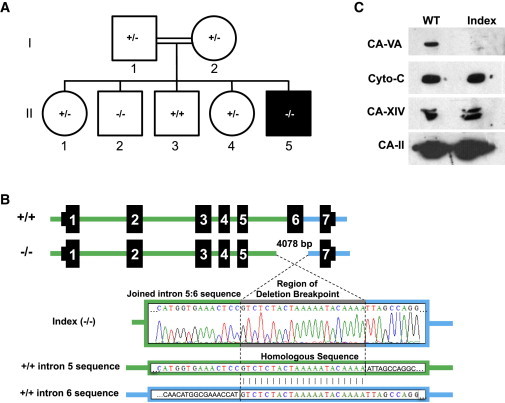
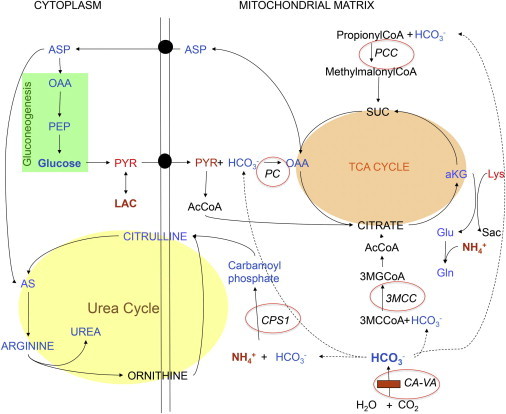
Four children in three unrelated families (one consanguineous) presented with lethargy, hyperlactatemia, and hyperammonemia of unexplained origin during the neonatal period and early childhood. We identified and validated three different CA5A alterations, including a homozygous missense mutation (c.697T>C) in two siblings, a homozygous splice site mutation (c.555G>A) leading to skipping of exon 4, and a homozygous 4 kb deletion of exon 6. The deleterious nature of the homozygous mutation c.697T>C (p.Ser233Pro) was demonstrated by reduced enzymatic activity and increased temperature sensitivity. Carbonic anhydrase VA (CA-VA) was absent in liver in the child with the homozygous exon 6 deletion. The metabolite profiles in the affected individuals fit CA-VA deficiency, showing evidence of impaired provision of bicarbonate to the four enzymes that participate in key pathways in intermediary metabolism: carbamoylphosphate synthetase 1 (urea cycle), pyruvate carboxylase (anaplerosis, gluconeogenesis), propionyl-CoA carboxylase, and 3-methylcrotonyl-CoA carboxylase (branched chain amino acids catabolism). In the three children who were administered carglumic acid, hyperammonemia resolved. CA-VA deficiency should therefore be added to urea cycle defects, organic acidurias, and pyruvate carboxylase deficiency as a treatable condition in the differential diagnosis of hyperammonemia in the neonate and young child.
Copyright © 2014 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Häberle J. Clinical and biochemical aspects of primary and secondary hyperammonemic disorders. Arch. Biochem. Biophys. 2013;536:101–108. - PubMed
-
- Nagao Y., Batanian J.R., Clemente M.F., Sly W.S. Genomic organization of the human gene (CA5) and pseudogene for mitochondrial carbonic anhydrase V and their localization to chromosomes 16q and 16p. Genomics. 1995;28:477–484. - PubMed
-
- Krebs J.F., Fierke C.A. Determinants of catalytic activity and stability of carbonic anhydrase II as revealed by random mutagenesis. J. Biol. Chem. 1993;268:948–954. - PubMed
-
- Andresen B.S., Dobrowolski S.F., O’Reilly L., Muenzer J., McCandless S.E., Frazier D.M., Udvari S., Bross P., Knudsen I., Banas R. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am. J. Hum. Genet. 2001;68:1408–1418. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
