

A penalized-likelihood method to estimate the distribution of selection coefficients from phylogenetic data
- PMID: 24532780
- PMCID: PMC4012484
- DOI: 10.1534/genetics.114.162263
A penalized-likelihood method to estimate the distribution of selection coefficients from phylogenetic data
Abstract
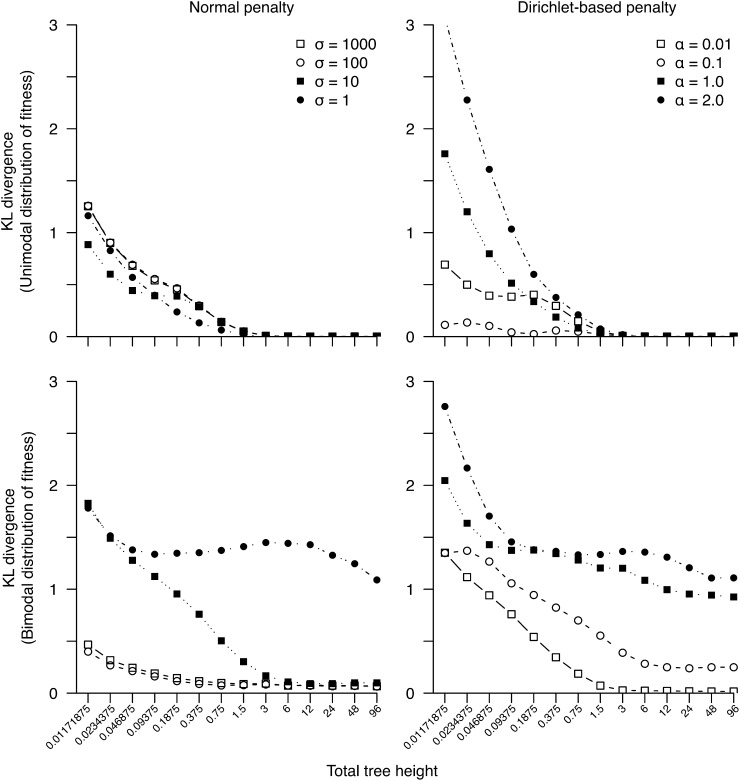
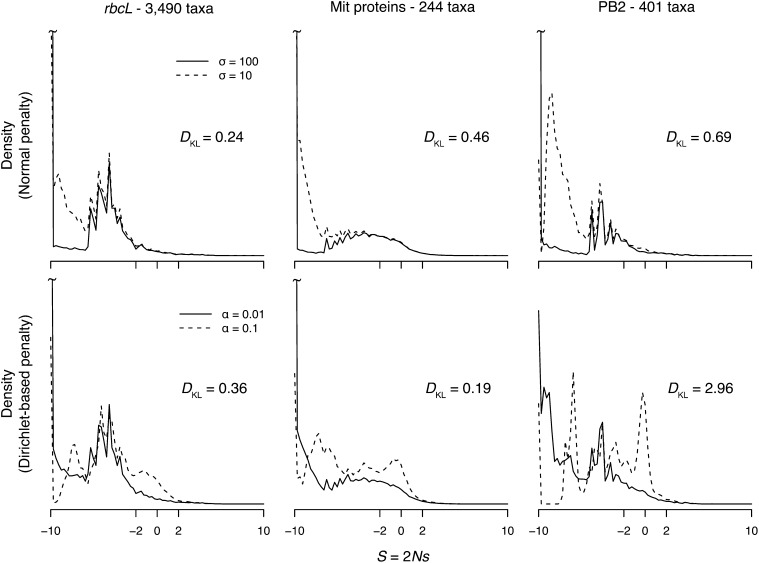
We develop a maximum penalized-likelihood (MPL) method to estimate the fitnesses of amino acids and the distribution of selection coefficients (S = 2Ns) in protein-coding genes from phylogenetic data. This improves on a previous maximum-likelihood method. Various penalty functions are used to penalize extreme estimates of the fitnesses, thus correcting overfitting by the previous method. Using a combination of computer simulation and real data analysis, we evaluate the effect of the various penalties on the estimation of the fitnesses and the distribution of S. We show the new method regularizes the estimates of the fitnesses for small, relatively uninformative data sets, but it can still recover the large proportion of deleterious mutations when present in simulated data. Computer simulations indicate that as the number of taxa in the phylogeny or the level of sequence divergence increases, the distribution of S can be more accurately estimated. Furthermore, the strength of the penalty can be varied to study how informative a particular data set is about the distribution of S. We analyze three protein-coding genes (the chloroplast rubisco protein, mammal mitochondrial proteins, and an influenza virus polymerase) and show the new method recovers a large proportion of deleterious mutations in these data, even under strong penalties, confirming the distribution of S is bimodal in these real data. We recommend the use of the new MPL approach for the estimation of the distribution of S in species phylogenies of protein-coding genes.
Keywords: chloroplast; fitness effects; influenza; mitochondria; penalized likelihood; selection coefficient.
Figures
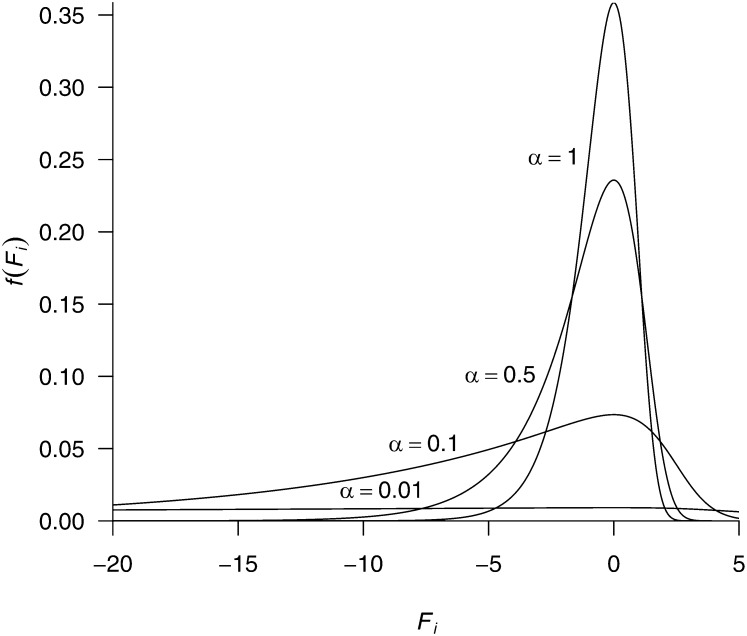
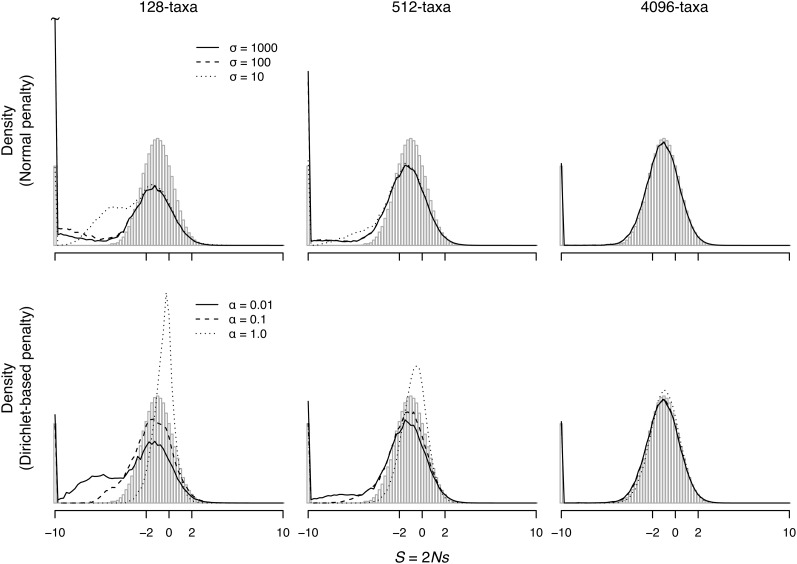
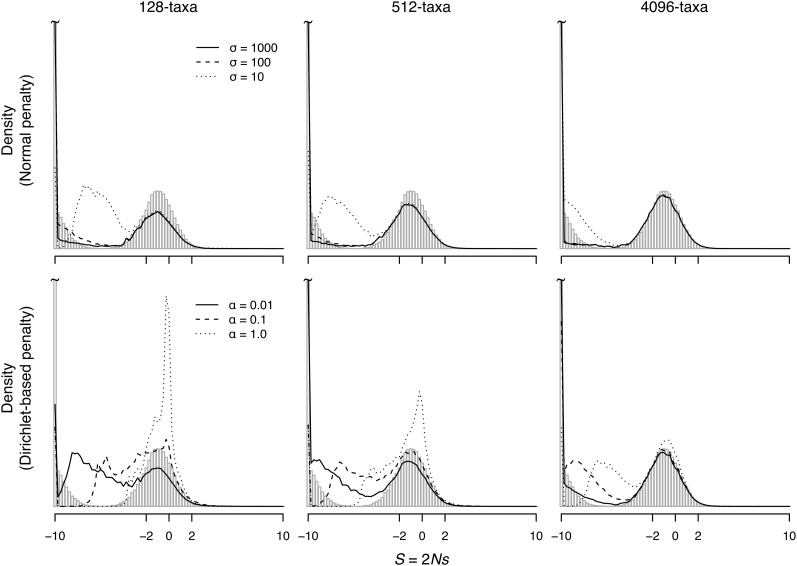
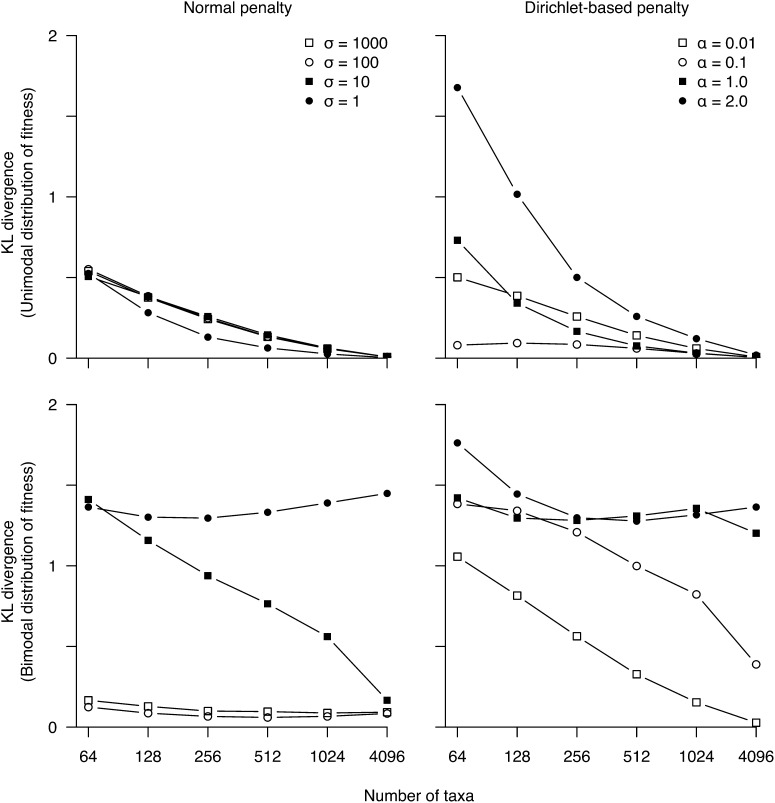
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
