

Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation
- PMID: 24532802
- PMCID: PMC3979407
- DOI: 10.1074/jbc.M113.531202
Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation
Abstract
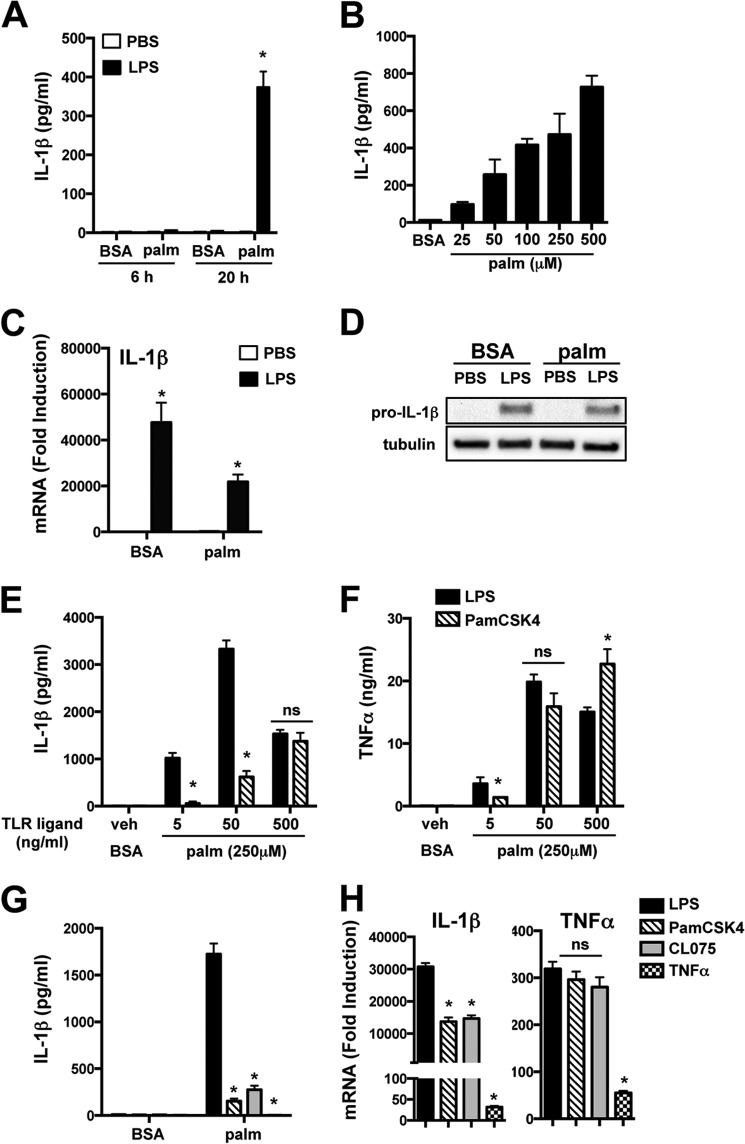
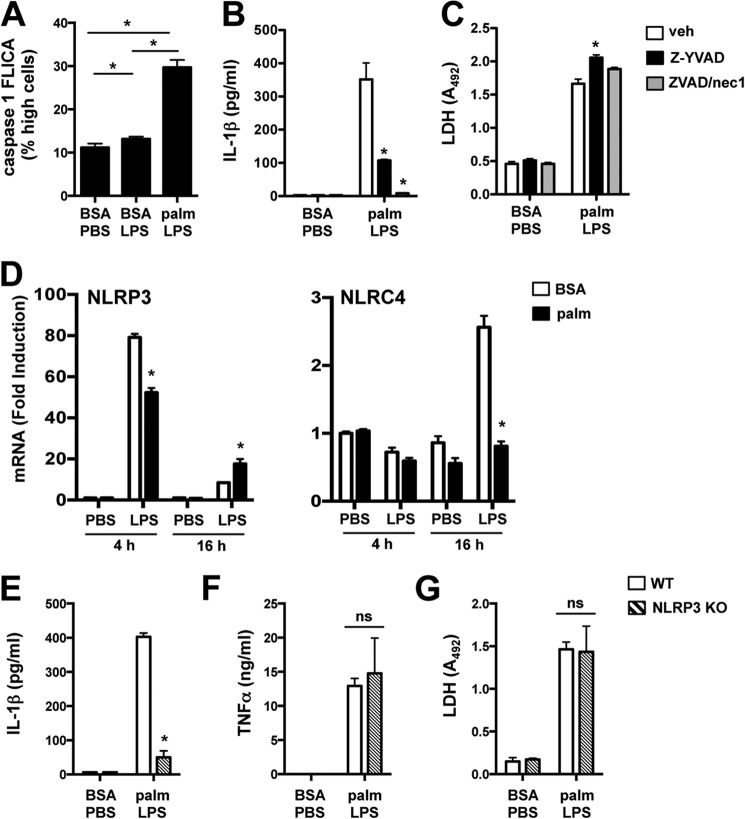
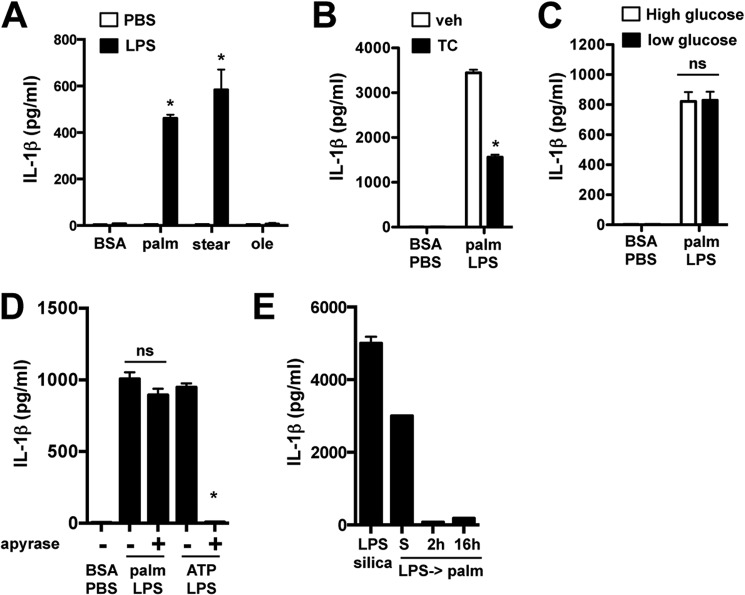
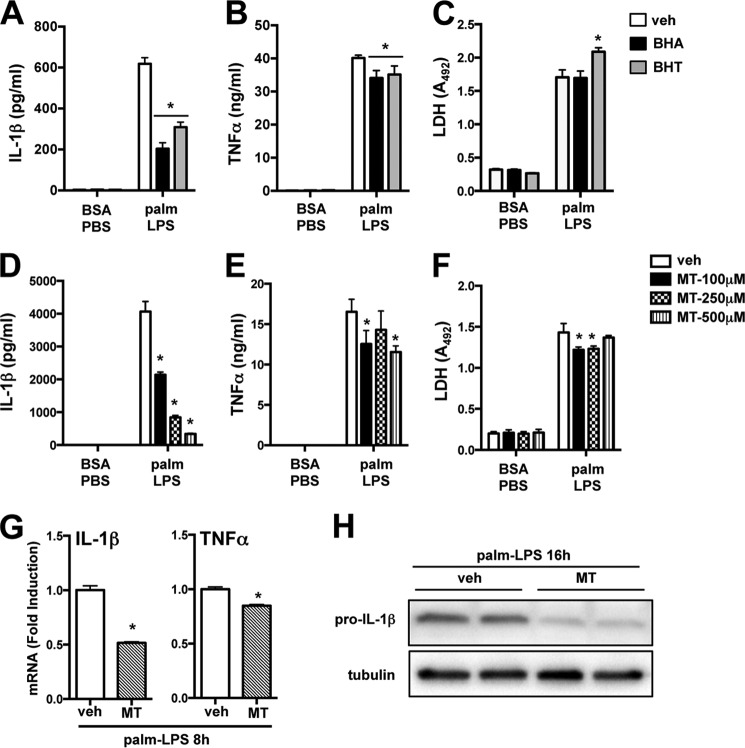
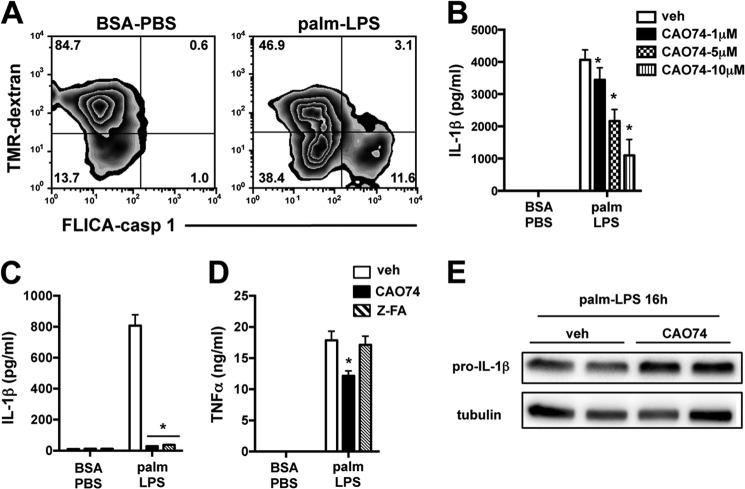
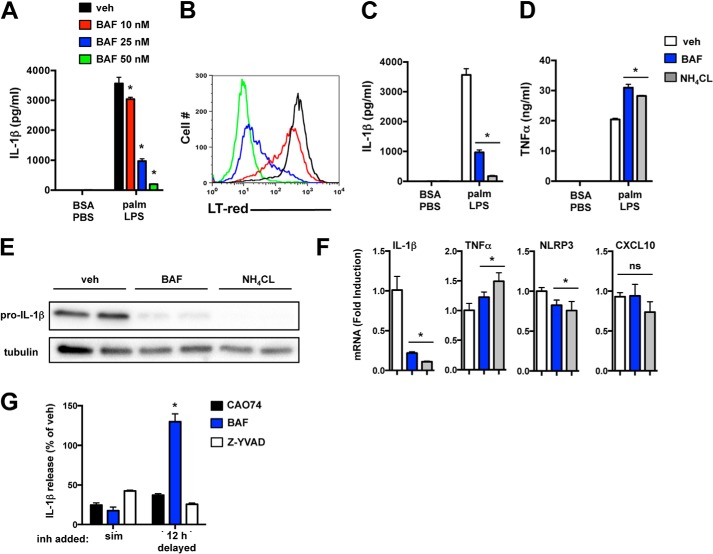
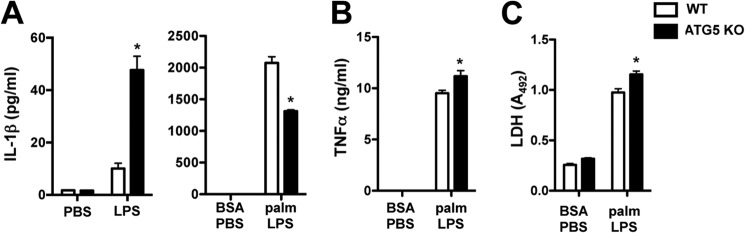
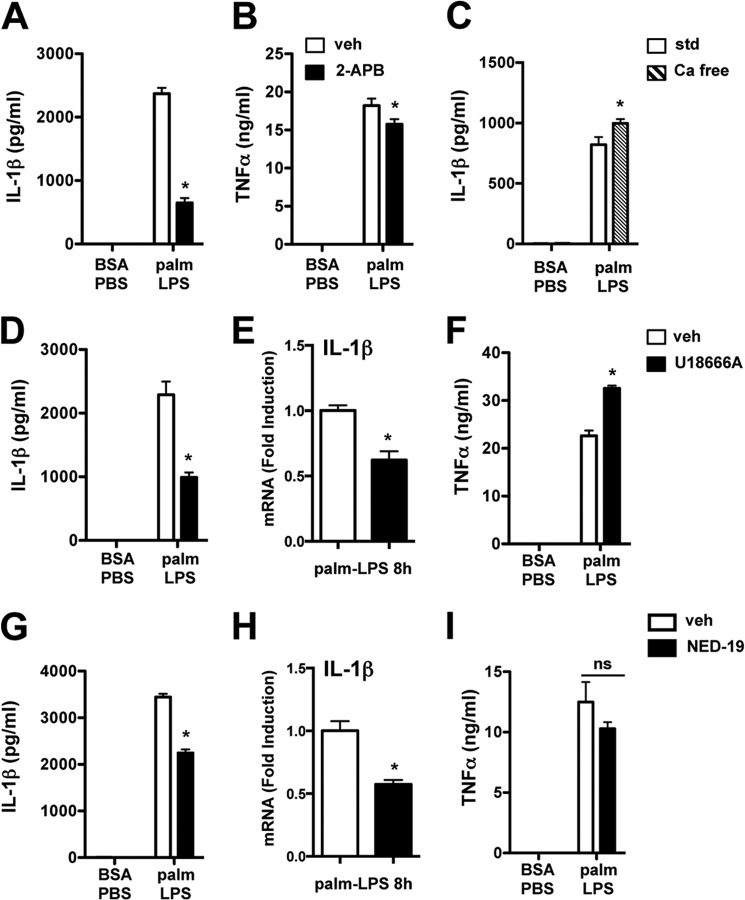
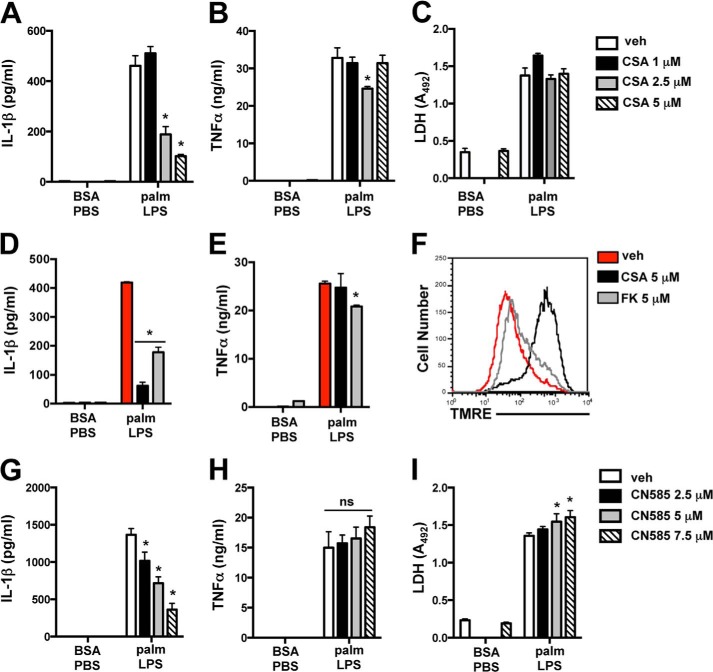
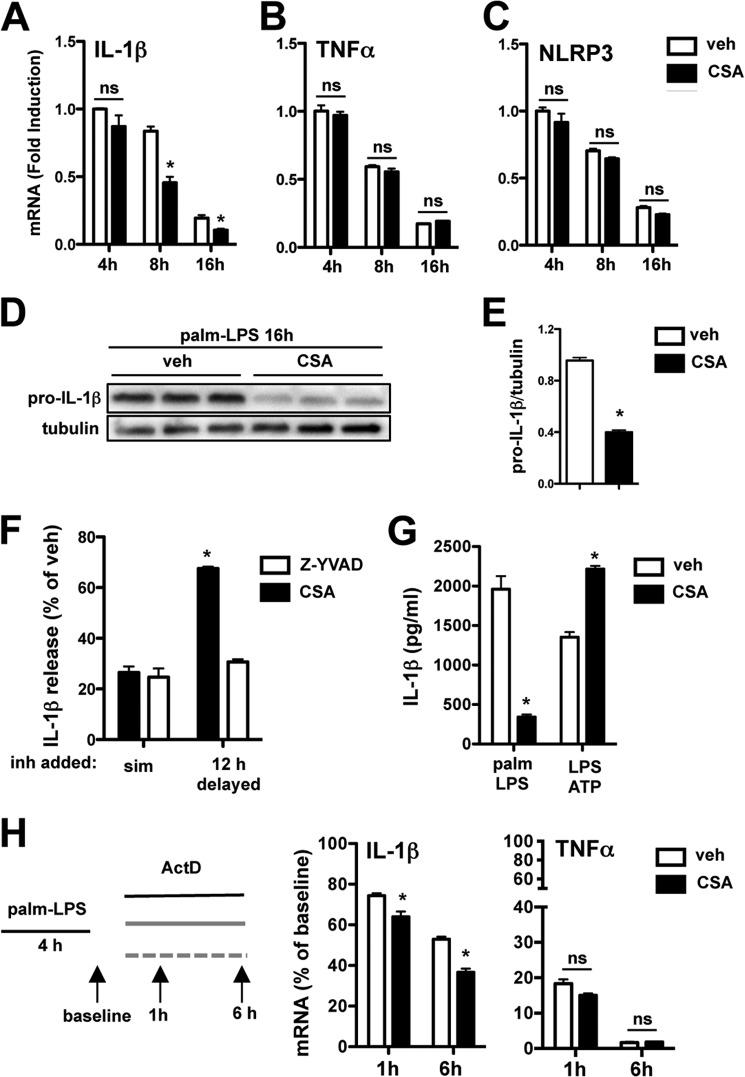
Macrophage dysfunction and inflammasome activation have been implicated in the pathogenesis of diabetes and its complications. Prolonged inflammation and impaired healing are hallmarks of the diabetic response to tissue injury, and excessive inflammasome activation has been associated in these phenotypes. However, the mechanisms that regulate the inflammasome in response to lipid metabolic and inflammatory stress are incompletely understood. We have shown previously that IL-1β secretion is induced in primary macrophages exposed to the dietary saturated fatty acid palmitate in combination with LPS. In this study, we sought to unravel the mechanisms underlying the activation of this lipotoxic inflammasome. We demonstrate that palmitate-loaded primary macrophages challenged with LPS activate the NLRP3 inflammasome through a mechanism that involves the lysosome. Interestingly, the lysosome was involved in both the regulation of pro-IL-1β levels and its subsequent cleavage/release. The lysosomal protease cathepsin B was required for IL-1β release but not pro-IL-1β production. In contrast, disrupting lysosomal calcium regulation decreased IL-1β release by reducing pro-IL-1β levels. The calcium pathway involved the calcium-activated phosphatase calcineurin, which stabilized IL-1β mRNA. Our findings provide evidence that the lysosome plays a key role in both the priming and assembly phases of the lipostoxic inflammasome. These findings have potential relevance to the hyperinflammatory phenotypes observed in diabetics during tissue damage or infection and identify lysosomes and calcineurin as potential therapeutic targets.
Keywords: Calcineurin; Diabetes; Inflammation; Lipids; Lysosomes; Macrophages.
Figures
References
-
- Mirza R., Koh T. J. (2011) Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 56, 256–264 - PubMed
-
- Kanter J. E., Kramer F., Barnhart S., Averill M. M., Vivekanandan-Giri A., Vickery T., Li L. O., Becker L., Yuan W., Chait A., Braun K. R., Potter-Perigo S., Sanda S., Wight T. N., Pennathur S., Serhan C. N., Heinecke J. W., Coleman R. A., Bornfeldt K. E. (2012) Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc. Natl. Acad. Sci. U.S.A. 109, E715–E724 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
