

Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer
- PMID: 24550319
- PMCID: PMC3932924
- DOI: 10.1073/pnas.1320956111
Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer
Abstract
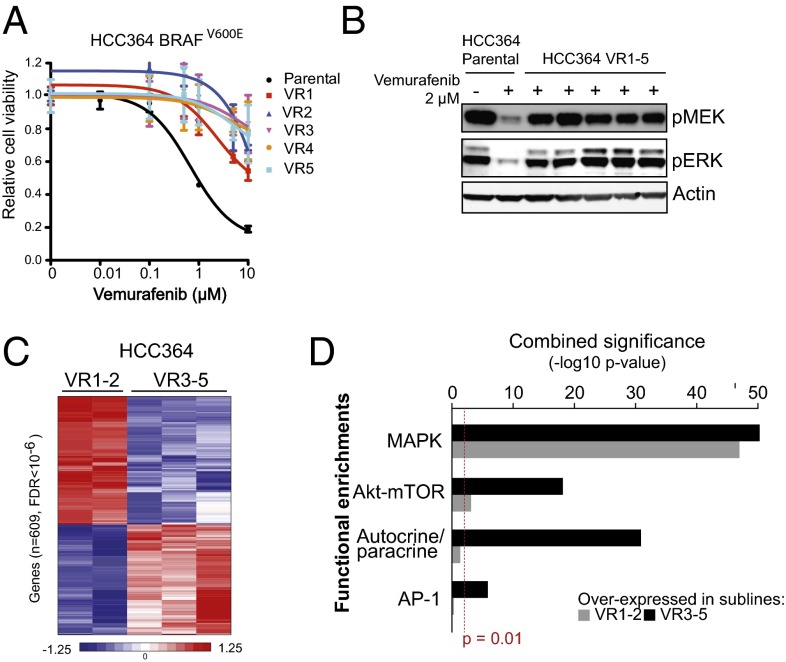
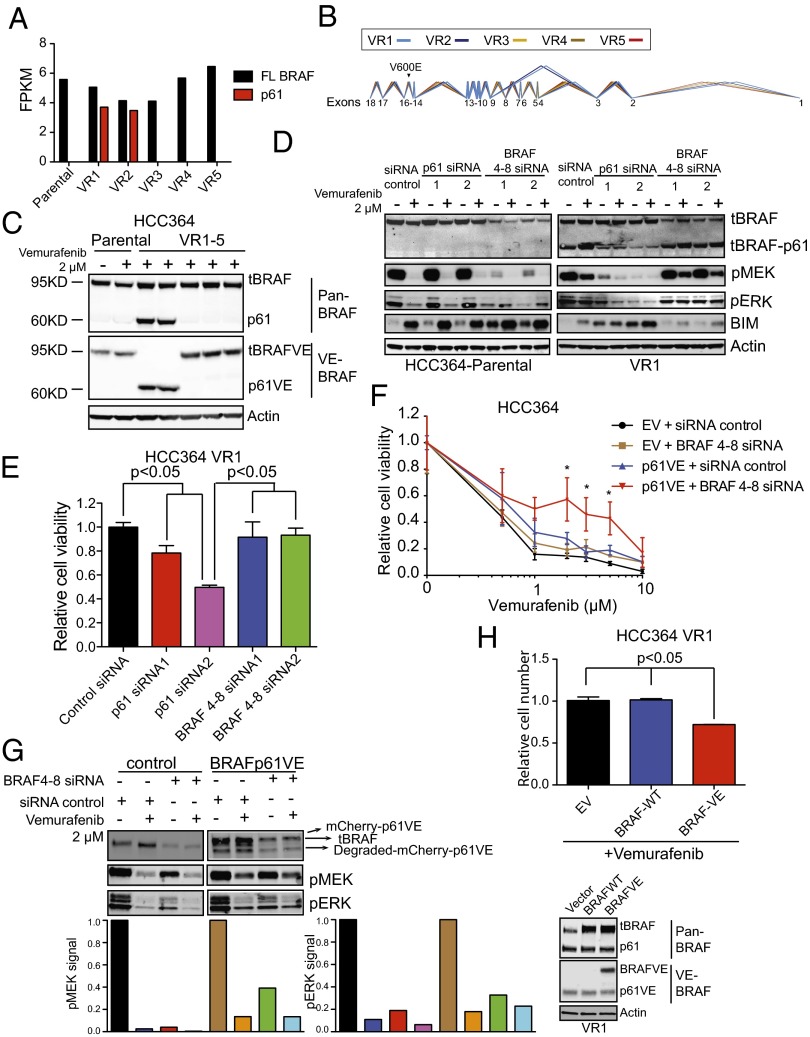
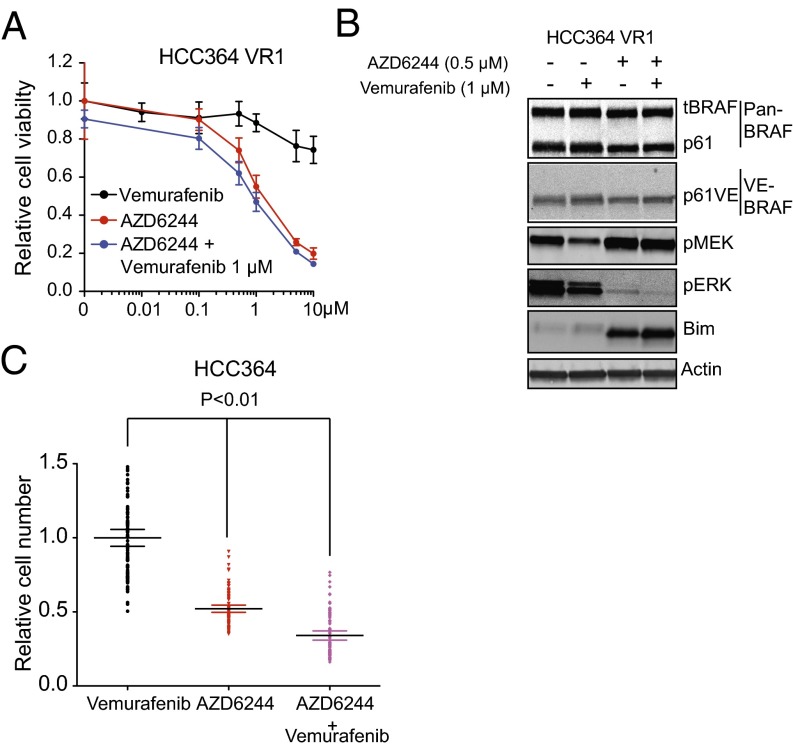
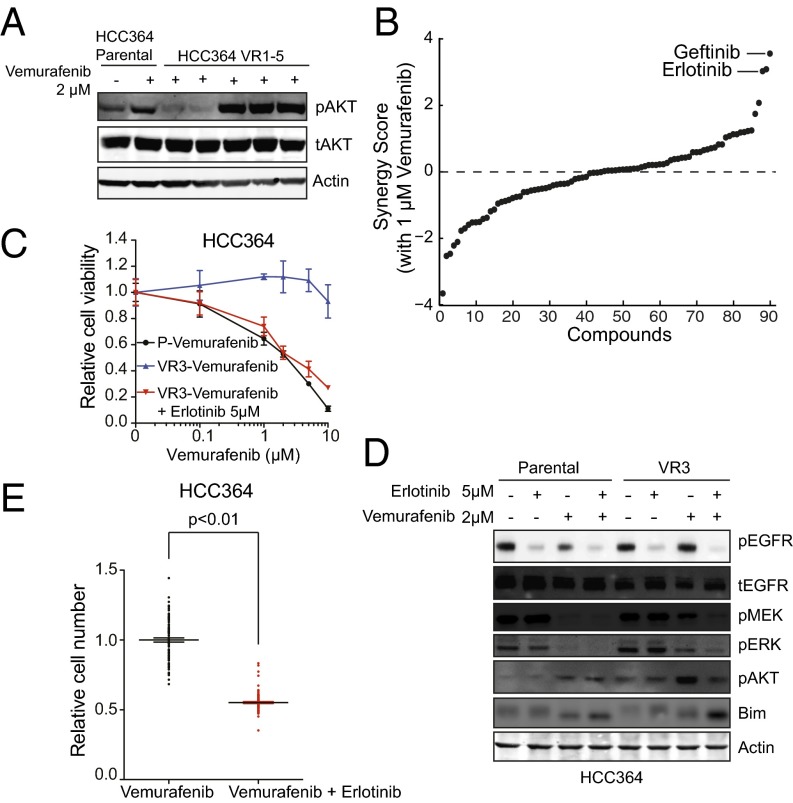
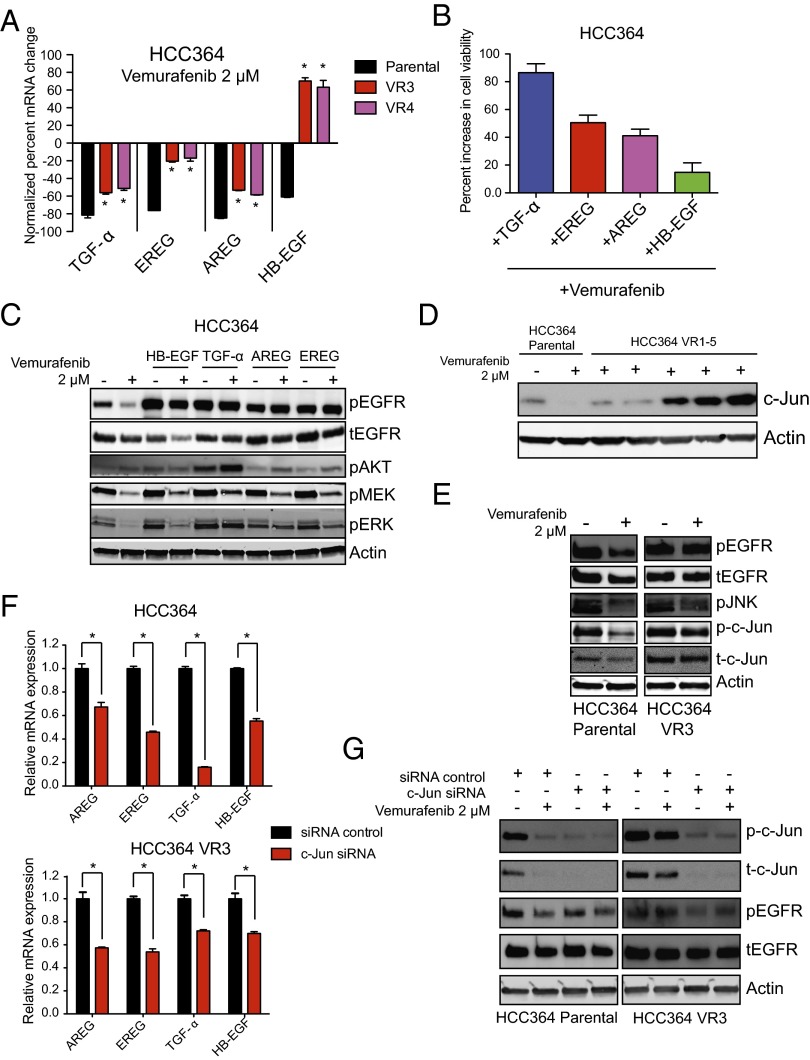
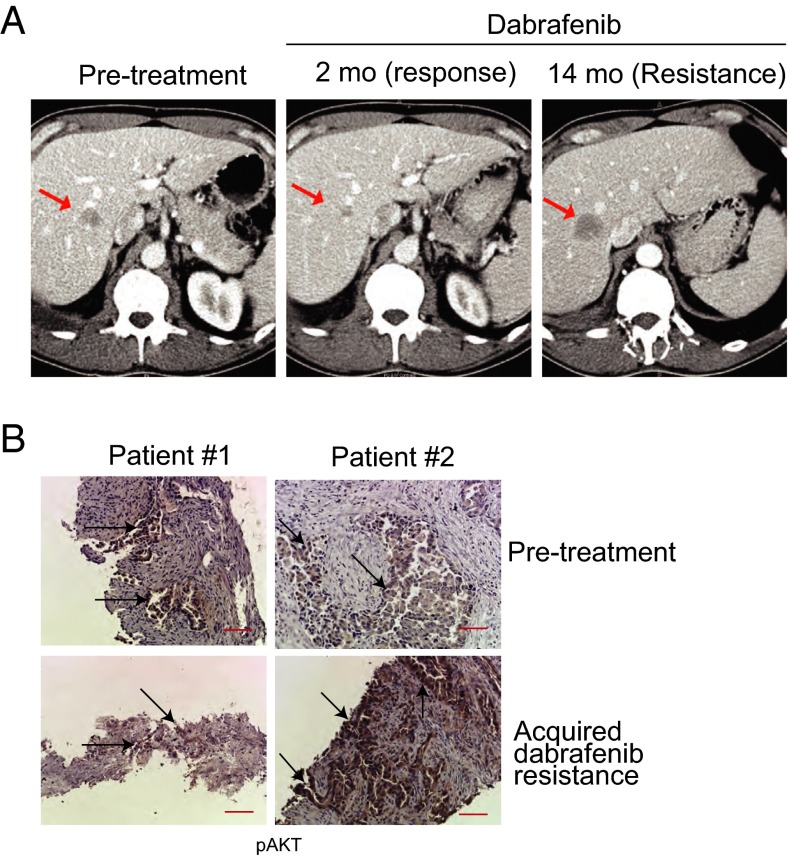
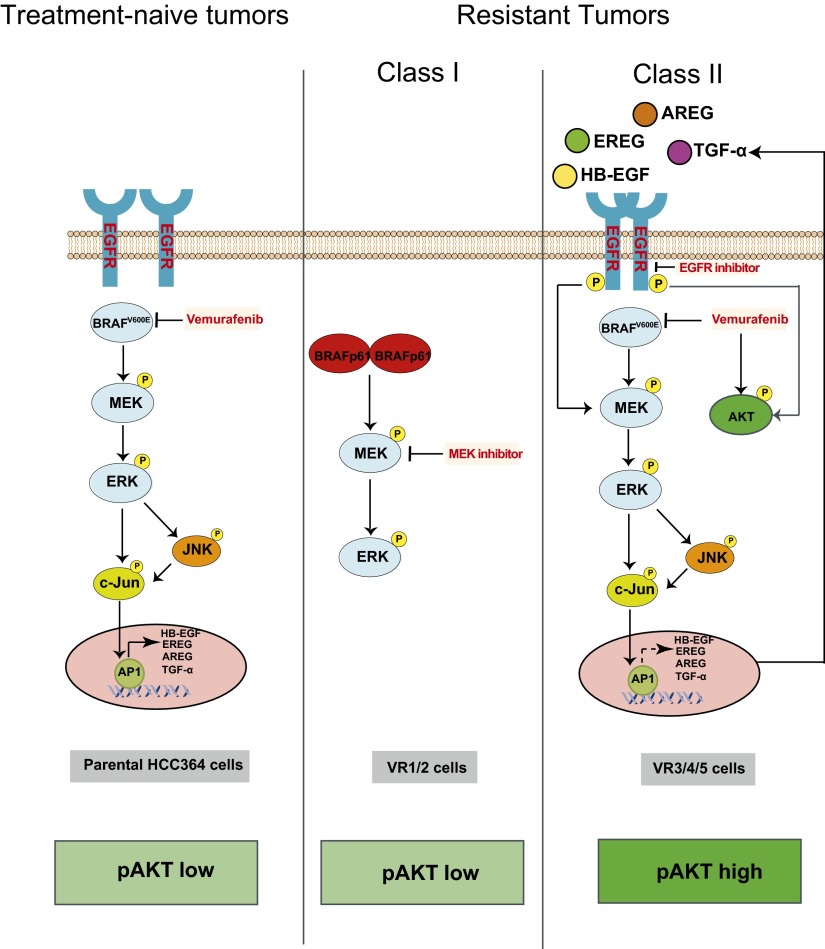
Oncogenic mutations in the BRAF kinase occur in 6-8% of nonsmall cell lung cancers (NSCLCs), accounting for more than 90,000 deaths annually worldwide. The biological and clinical relevance of these BRAF mutations in NSCLC is incompletely understood. Here we demonstrate that human NSCLC cells with BRAF(V600E), but not other BRAF mutations, initially are sensitive to BRAF-inhibitor treatment. However, these BRAF(V600E) NSCLC cells rapidly acquire resistance to BRAF inhibition through at least one of two discrete molecular mechanisms: (i) loss of full-length BRAF(V600E) coupled with expression of an aberrant form of BRAF(V600E) that retains RAF pathway dependence or (ii) constitutive autocrine EGF receptor (EGFR) signaling driven by c-Jun-mediated EGFR ligand expression. BRAF(V600E) cells with EGFR-driven resistance are characterized by hyperphosphorylated protein kinase AKT, a biomarker we validated in BRAF inhibitor-resistant NSCLC clinical specimens. These data reveal the multifaceted molecular mechanisms by which NSCLCs establish and regulate BRAF oncogene dependence, provide insights into BRAF-EGFR signaling crosstalk, and uncover mechanism-based strategies to optimize clinical responses to BRAF oncogene inhibition.
Keywords: combination therapy; targeted therapy.
Conflict of interest statement
The authors declare a conflict of interest. Gideon Bollag is an employee of Plexxikon Inc., which owns and markets vemurafenib.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
