

Requirement for TLR2 in the development of albuminuria, inflammation and fibrosis in experimental diabetic nephropathy
- PMID: 24551269
- PMCID: PMC3925893
Requirement for TLR2 in the development of albuminuria, inflammation and fibrosis in experimental diabetic nephropathy
Abstract
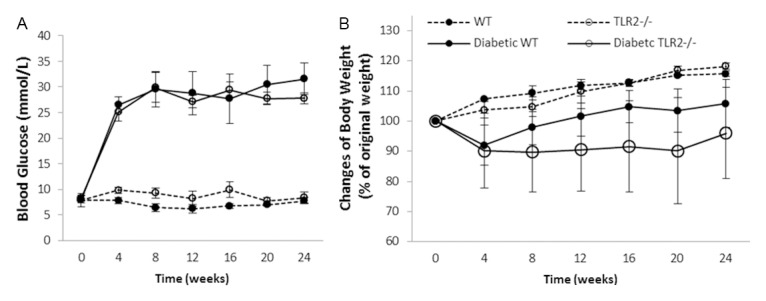
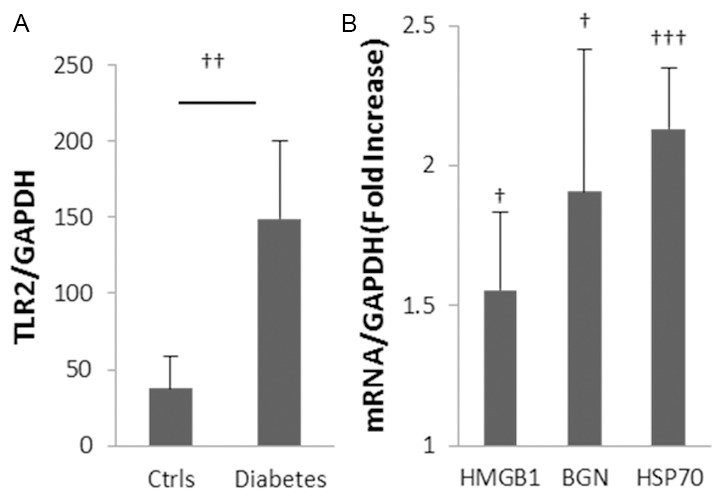
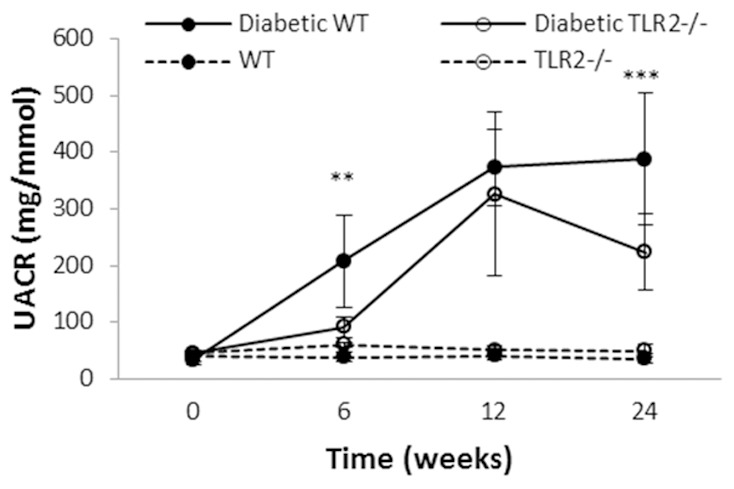
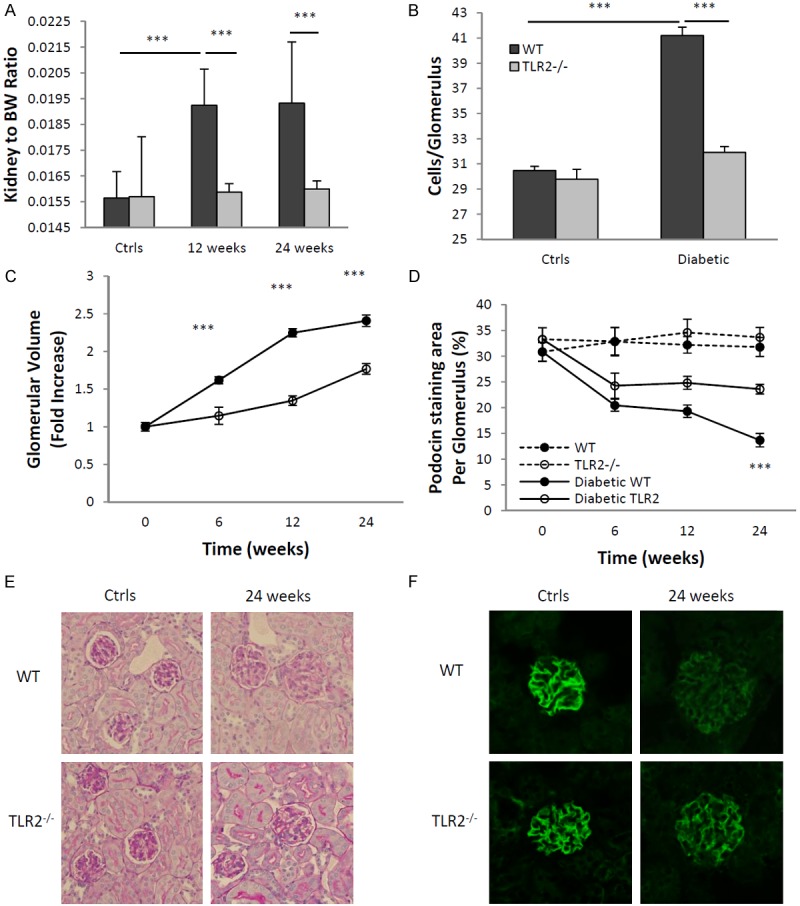
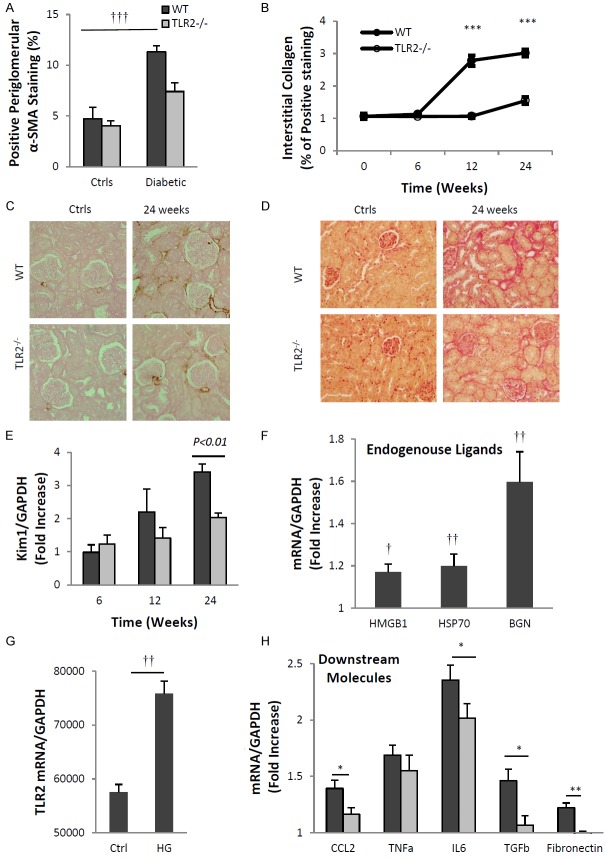
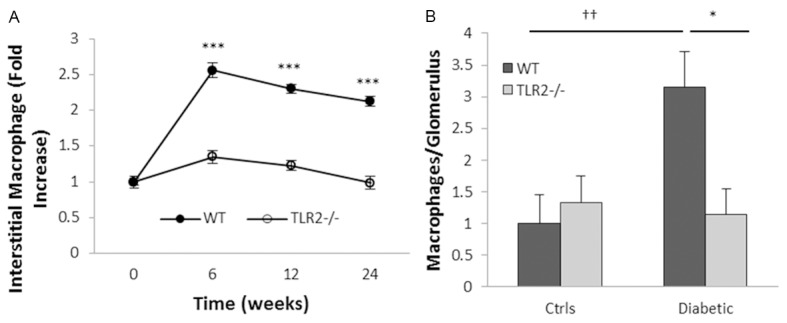
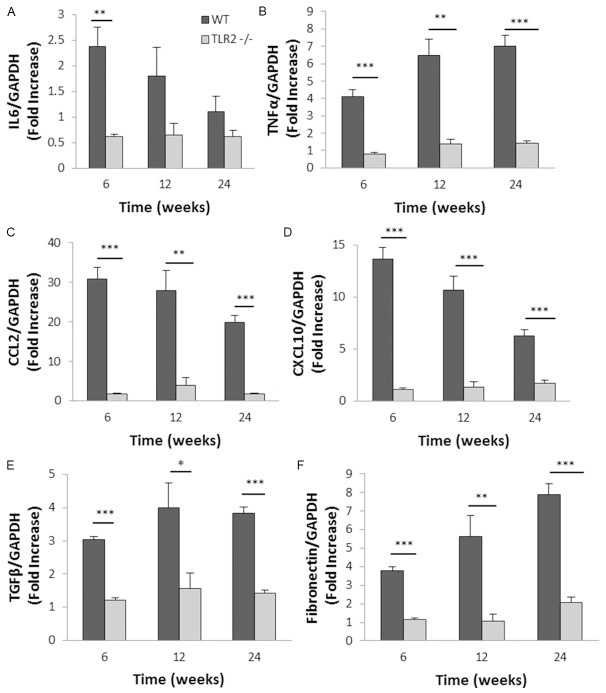
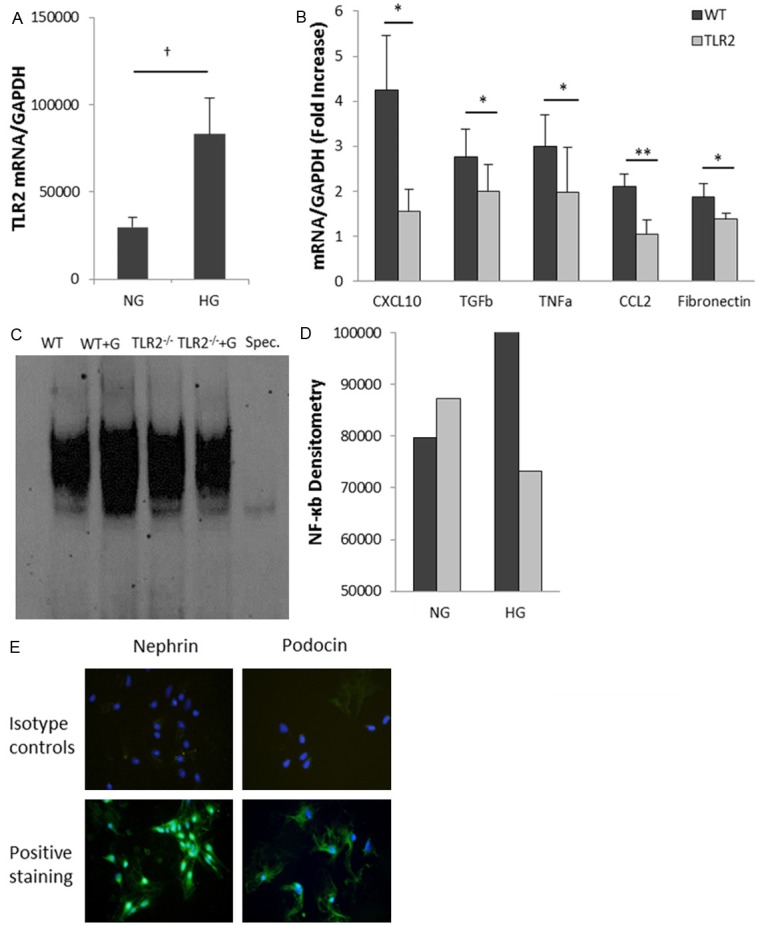
Inflammation and fibrosis are essential elements of diabetic nephropathy (DN). We tested the hypothesis that these elements are dependent upon Toll-like receptor 2 (TLR2) signalling by examining WT and TLR2(-/-) mice in an experimental model of DN. Diabetes was induced in WT and TLR2(-/-) mice by i.p. injection of streptozotocin. Kidney injury was assessed at 6, 12 and 24 weeks after induction of diabetes. Gene expression of TLR2, its endogenous ligands and downstream cytokines, chemokines and fibrogenic molecules were upregulated in kidneys from WT mice with streptozotocin diabetes. TLR2(-/-) mice were protected against the development of DN, exhibiting less albuminuria, inflammation, glomerular hypertrophy and hypercellularity, podocyte and tubular injury as compared to diabetic WT controls. Marked reductions in interstitial collagen deposition, myofibroblast activation (α-SMA) and expression of fibrogenic genes (TGF-β and fibronectin) were also evident in TLR2 deficient mice. Consistent with our in vivo results, high glucose directly promoted TLR2 activation in podocytes and tubular epithelial cells (TECs) in vitro, resulting in NF-κB activation, inflammation and TGF-β production. We conclude that TLR2 was required for the full development of inflammation, kidney damage and fibrosis in this model of DN. As TLR2 is known to be expressed by intrinsic kidney cells and as high concentration glucose stimulated podocytes and TECs in vitro to express TLR2 and TLR2 ligands, pro-inflammatory and pro-fibrotic cytokines in a TLR2 dependent manner in the present study, it appears likely that TLR2 signalling in intrinsic kidney cells contributes to the pathogenesis of diabetic nephropathy.
Keywords: Diabetic nephropathy; Toll like receptor 2; fibrosis; podocytes.
Figures
References
-
- Hovind P, Tarnow L, Rossing K, Rossing P, Eising S, Larsen N, Binder C, Parving HH. Decreasing incidence of severe diabetic microangiopathy in type 1 diabetes. Diabetes Care. 2003;26:1258–1264. - PubMed
-
- White SL, Cass A, Atkins RC, Chadban SJ. Chronic kidney disease in the general population. Adv Chronic Kidney Dis. 2005;12:5–13. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials