

NOX1 is responsible for cell death through STAT3 activation in hyperoxia and is associated with the pathogenesis of acute respiratory distress syndrome
- PMID: 24551274
- PMCID: PMC3925898
NOX1 is responsible for cell death through STAT3 activation in hyperoxia and is associated with the pathogenesis of acute respiratory distress syndrome
Abstract
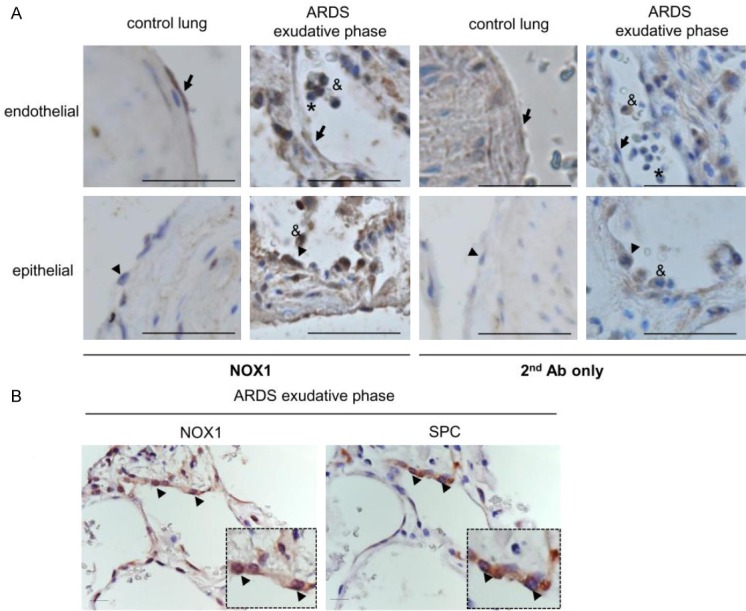
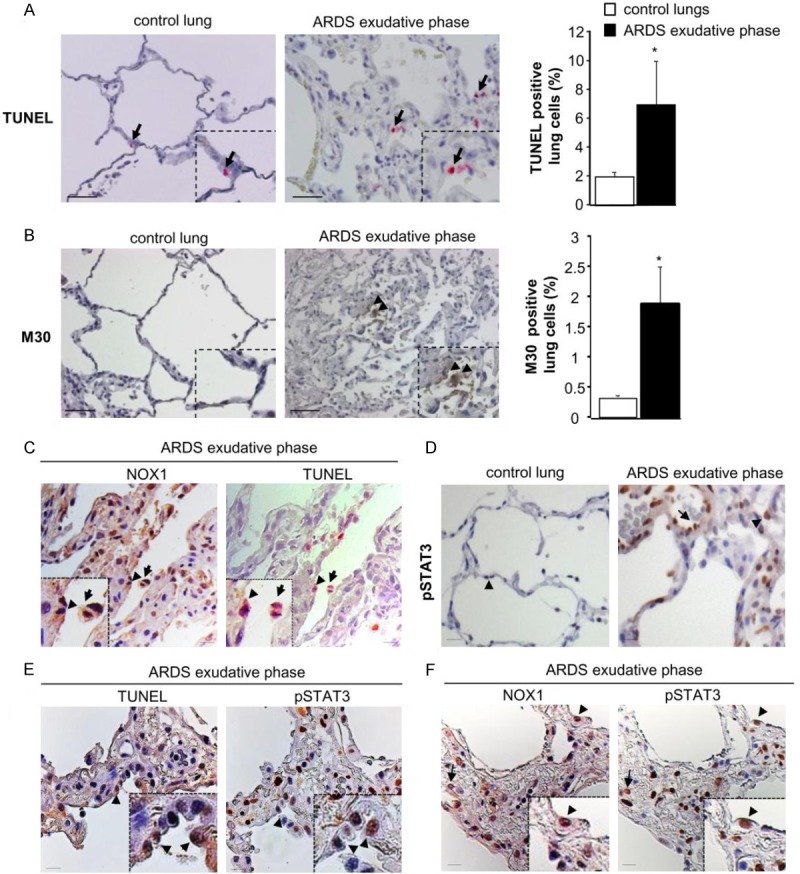
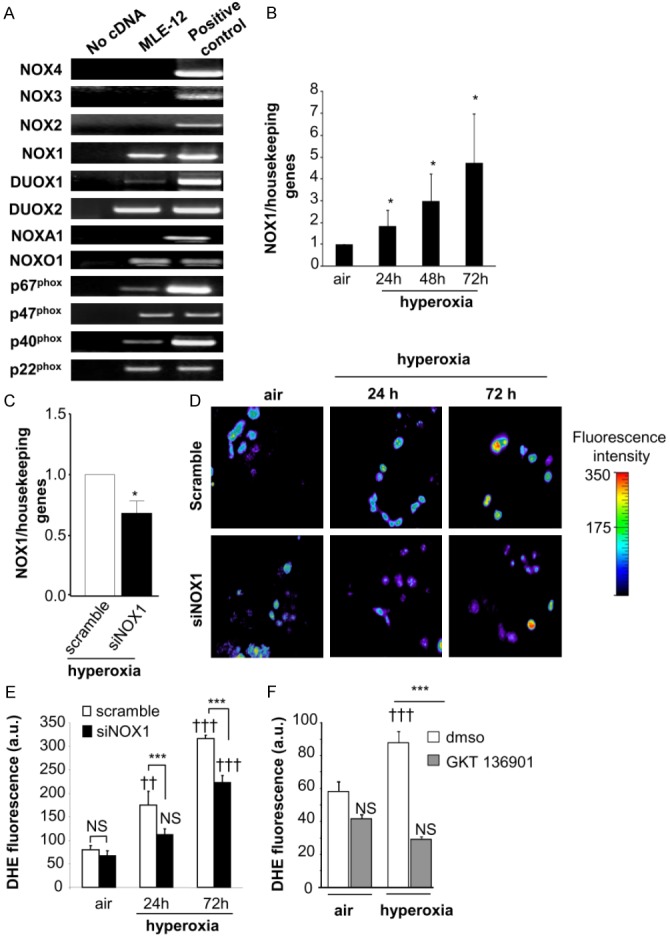
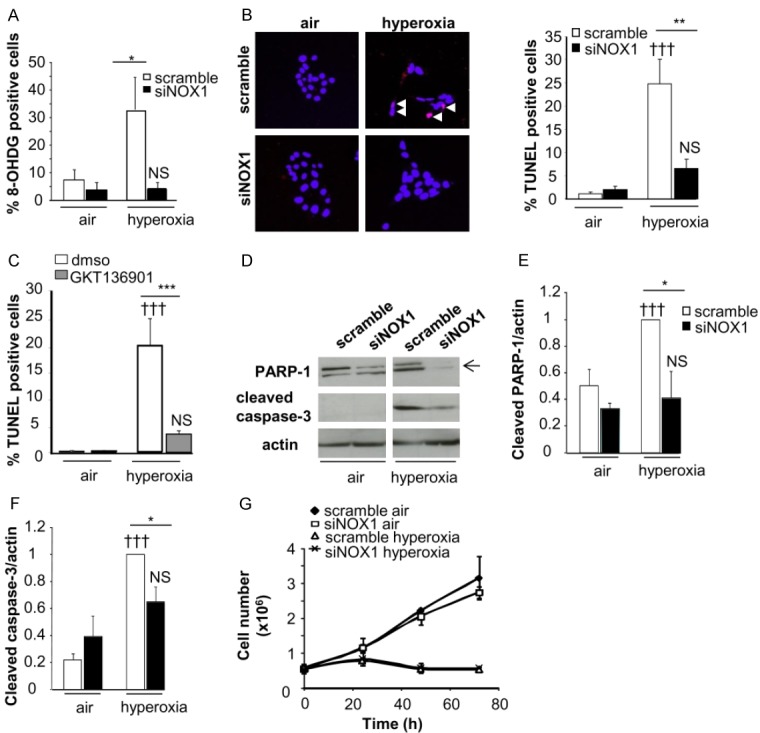
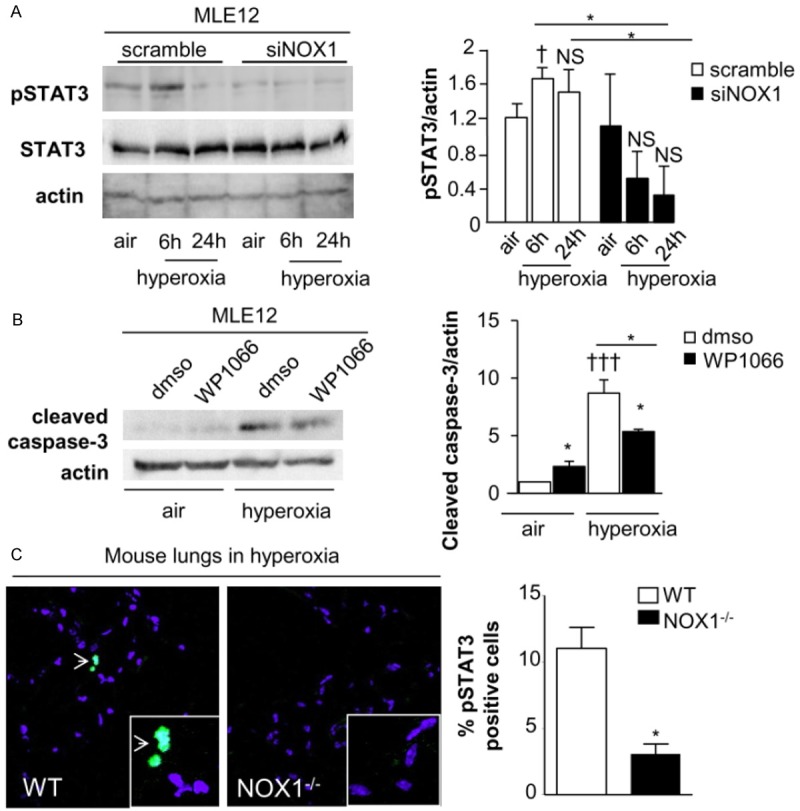
Reactive oxygen species (ROS) contribute to alveolar cell death in acute respiratory distress syndrome (ARDS) and we previously demonstrated that NOX1-derived ROS contributed to hyperoxia-induced alveolar cell death in mice. The study investigates whether NOX1 expression is modulated in epithelial cells concomitantly to cell death and associated to STAT3 signaling in the exudative phase of ARDS. In addition, the role of STAT3 activation in NOX1-dependent epithelial cell death was confirmed by using a lung epithelial cell line and in mice exposed to hyperoxia. NOX1 expression, cell death and STAT3 staining were evaluated in the lungs of control and ARDS patients by immunohistochemistry. In parallel, a stable NOX1-silenced murine epithelial cell line (MLE12) and NOX1-deficient mice were used to characterize signalling pathways. In the present study, we show that NOX1 is detected in alveolar epithelial cells of ARDS patients in the exudative stage. In addition, increased alveolar epithelial cell death and phosphorylated STAT3 are observed in ARDS patients and associated with NOX1 expression. Phosphorylated STAT3 is also correlated with TUNEL staining. We also confirmed that NOX1-dependent STAT3 activation participates to alveolar epithelial cell death. Silencing and acute inhibition of NOX1 in MLE12 led to decreased cell death and cleaved-caspase 3 induced by hyperoxia. Additionally, hyperoxia-induced STAT3 phosphorylation is dependent on NOX1 expression and associated with cell death in MLE12 and mice. This study demonstrates that NOX1 is involved in human ARDS pathophysiology and is responsible for the damage occurring in alveolar epithelial cells at least in part via STAT3 signalling pathways.
Keywords: ARDS; NOX1; ROS; STAT3; cell death; hyperoxia.
Figures
References
-
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. - PubMed
-
- Lee KS, Choi YH, Kim YS, Baik SH, Oh YJ, Sheen SS, Park JH, Hwang SC, Park KJ. Evaluation of bronchoalveolar lavage fluid from ARDS patients with regard to apoptosis. Respir Med. 2008;102:464–469. - PubMed
-
- Lamb NJ, Gutteridge JM, Baker C, Evans TW, Quinlan GJ. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit Care Med. 1999;27:1738–1744. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous