

N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury
- PMID: 24553937
- PMCID: PMC3929763
- DOI: 10.1523/JNEUROSCI.1948-13.2014
N-acetyl-serotonin offers neuroprotection through inhibiting mitochondrial death pathways and autophagic activation in experimental models of ischemic injury
Abstract
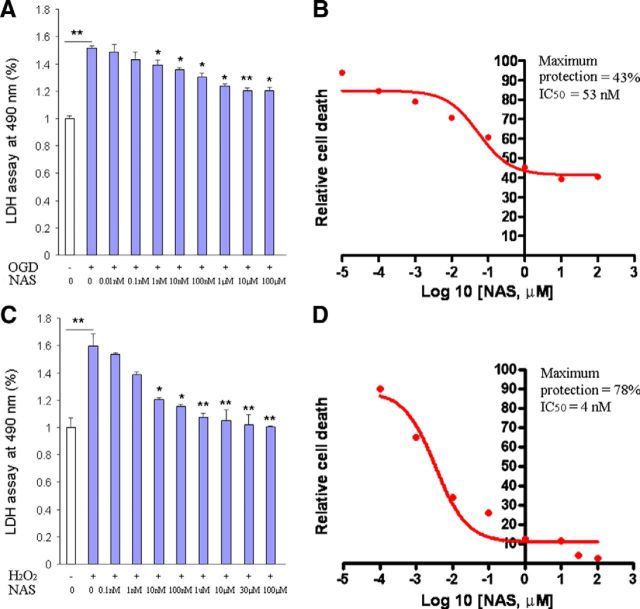
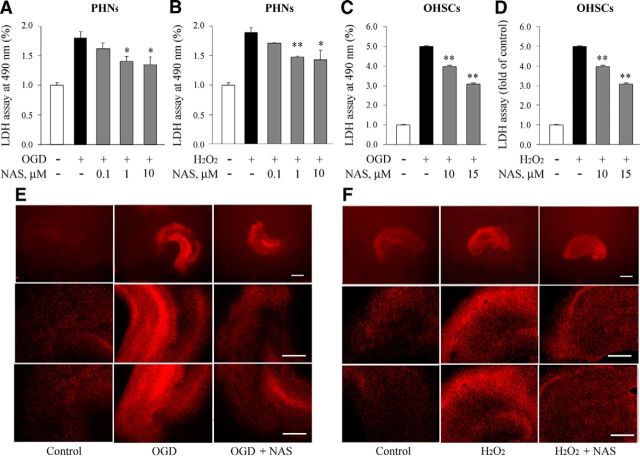
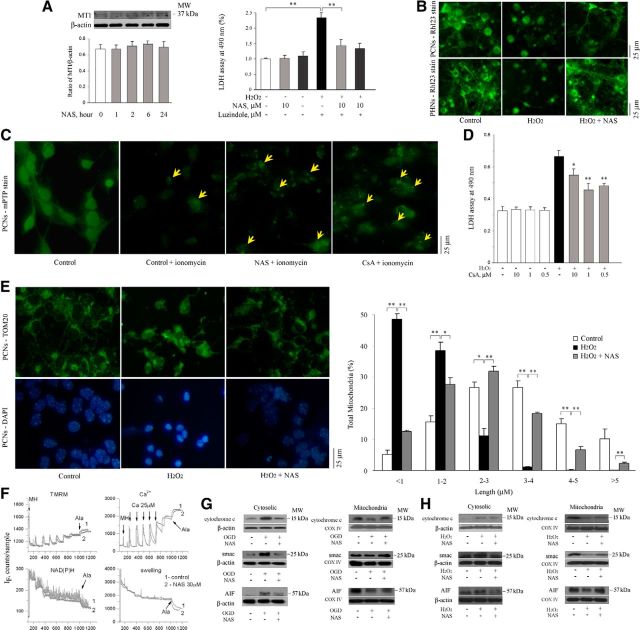
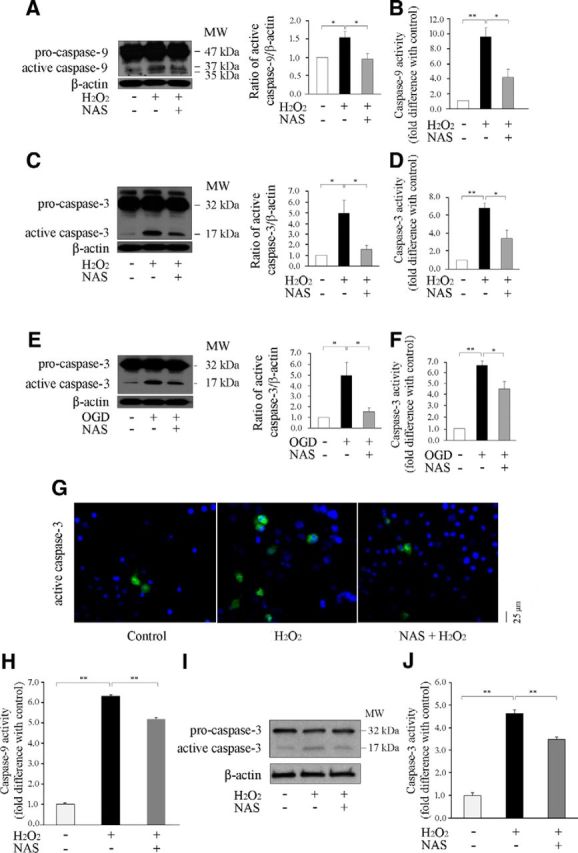
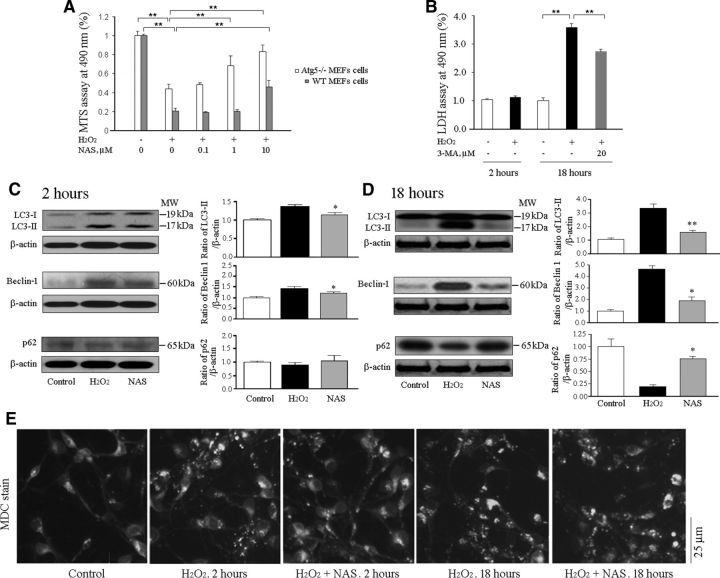
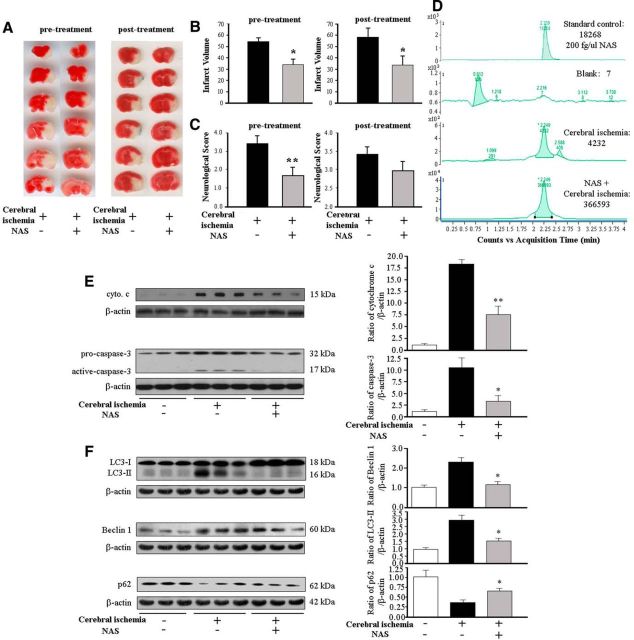
N-acetylserotonin (NAS) is an immediate precursor of melatonin, which we have reported is neuroprotective against ischemic injury. Here we test whether NAS is a potential neuroprotective agent in experimental models of ischemic injury. We demonstrate that NAS inhibits cell death induced by oxygen-glucose deprivation or H2O2 in primary cerebrocortical neurons and primary hippocampal neurons in vitro, and organotypic hippocampal slice cultures ex vivo and reduces hypoxia/ischemia injury in the middle cerebral artery occlusion mouse model of cerebral ischemia in vivo. We find that NAS is neuroprotective by inhibiting the mitochondrial cell death pathway and the autophagic cell death pathway. The neuroprotective effects of NAS may result from the influence of mitochondrial permeability transition pore opening, mitochondrial fragmentation, and inhibition of the subsequent release of apoptogenic factors cytochrome c, Smac, and apoptosis-inducing factor from mitochondria to cytoplasm, and activation of caspase-3, -9, as well as the suppression of the activation of autophagy under stress conditions by increasing LC3-II and Beclin-1 levels and decreasing p62 level. However, NAS, unlike melatonin, does not provide neuroprotection through the activation of melatonin receptor 1A. We demonstrate that NAS reaches the brain subsequent to intraperitoneal injection using liquid chromatography/mass spectrometry analysis. Given that it occurs naturally and has low toxicity, NAS, like melatonin, has potential as a novel therapy for ischemic injury.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials