

Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release
- PMID: 24554772
- PMCID: PMC3985066
- DOI: 10.4049/jimmunol.1302484
Cholesterol crystals induce complement-dependent inflammasome activation and cytokine release
Abstract
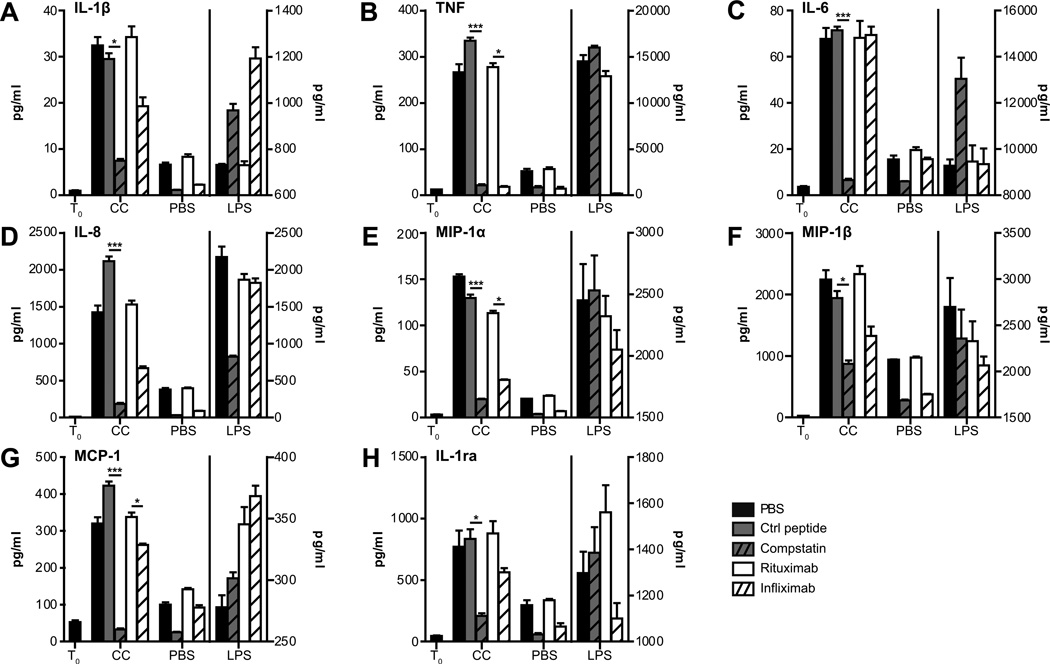
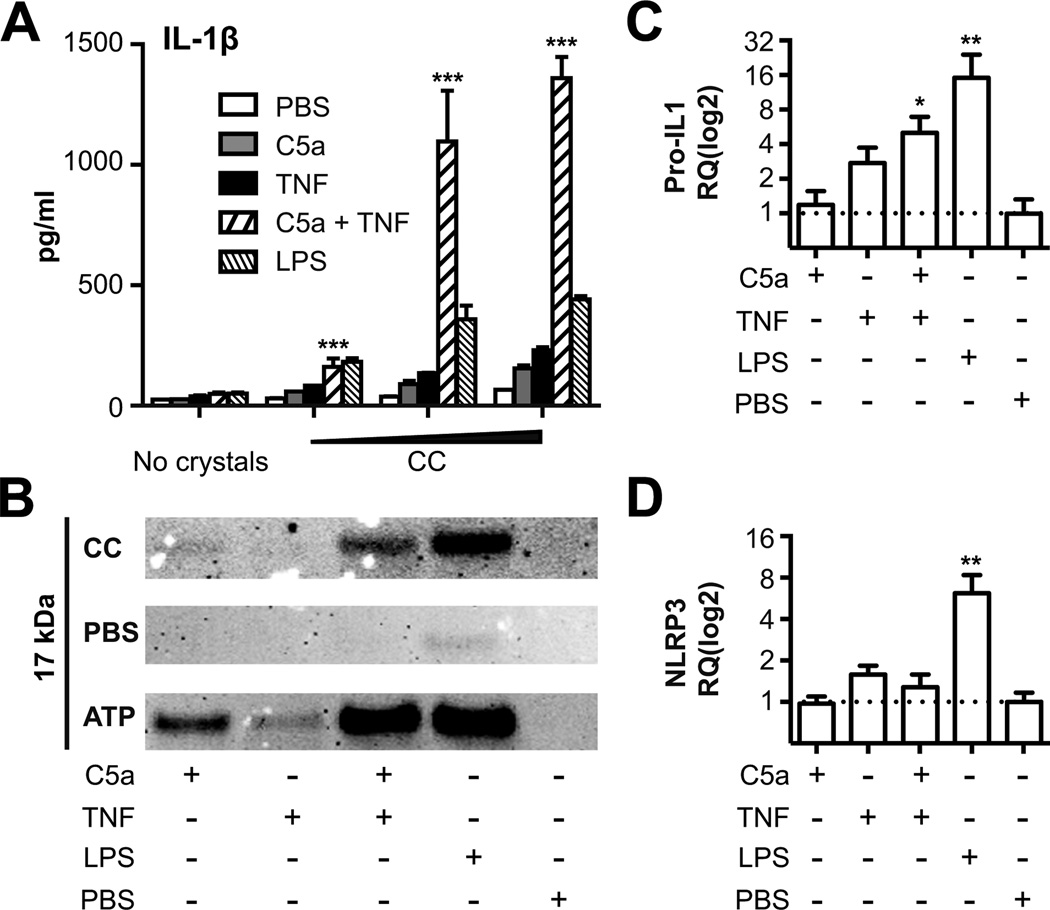
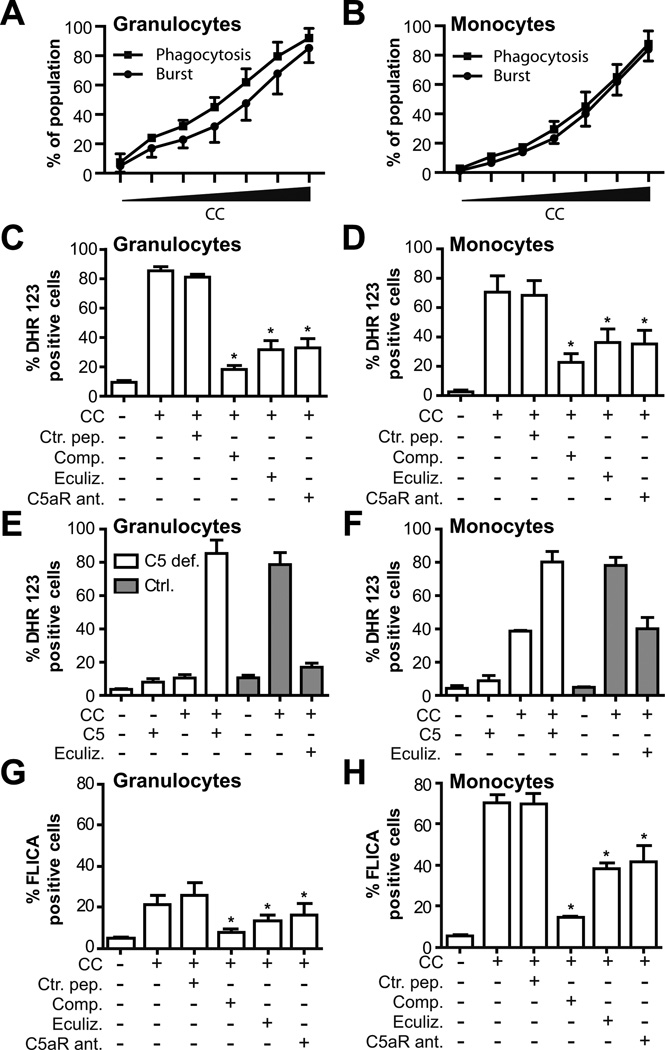
Inflammation is associated with development of atherosclerosis, and cholesterol crystals (CC) have long been recognized as a hallmark of atherosclerotic lesions. CC appear early in the atheroma development and trigger inflammation by NLRP3 inflammasome activation. In this study we hypothesized whether CC employ the complement system to activate inflammasome/caspase-1, leading to release of mature IL-1β, and whether complement activation regulates CC-induced cytokine production. In this study we describe that CC activated both the classical and alternative complement pathways, and C1q was found to be crucial for the activation. CC employed C5a in the release of a number of cytokines in whole blood, including IL-1β and TNF. CC induced minimal amounts of cytokines in C5-deficient whole blood, until reconstituted with C5. Furthermore, C5a and TNF in combination acted as a potent primer for CC-induced IL-1β release by increasing IL-1β transcripts. CC-induced complement activation resulted in upregulation of complement receptor 3 (CD11b/CD18), leading to phagocytosis of CC. Also, CC mounted a complement-dependent production of reactive oxygen species and active caspase-1. We conclude that CC employ the complement system to induce cytokines and activate the inflammasome/caspase-1 by regulating several cellular responses in human monocytes. In light of this, complement inhibition might be an interesting therapeutic approach for treatment of atherosclerosis.
Conflict of interest statement
Disclosures:
Conflict of interest: The co-author, Prof. J.D.L., is the holder of several patent applications on complement inhibitors and the founder of Amyndas Pharmaceuticals, which is developing complement inhibitors for clinical applications. The other authors report no conflict.
Figures




References
-
- Small DM. George Lyman Duff memorial lecture. Progression and regression of atherosclerotic lesions. Insights from lipid physical biochemistry. Arteriosclerosis. 1988;8:103–129. - PubMed
-
- Abela GS. Cholesterol crystals piercing the arterial plaque and intima trigger local and systemic inflammation. J. Clin. Lipidol. 2010;4:156–164. - PubMed
-
- Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W, Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler. Thromb. Vasc. Biol. 1995;15:1512–1531. - PubMed
-
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. - PMC - PubMed
-
- Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013;14:812–820. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
