

The MAPK-activated protein kinase 2 mediates gemcitabine sensitivity in pancreatic cancer cells
- PMID: 24556918
- PMCID: PMC3984311
- DOI: 10.4161/cc.28292
The MAPK-activated protein kinase 2 mediates gemcitabine sensitivity in pancreatic cancer cells
Abstract
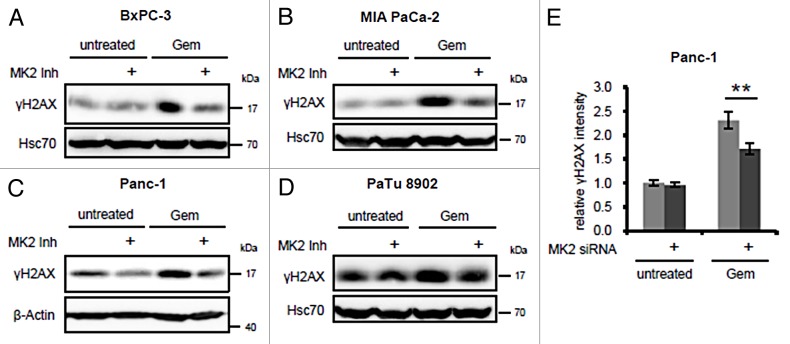
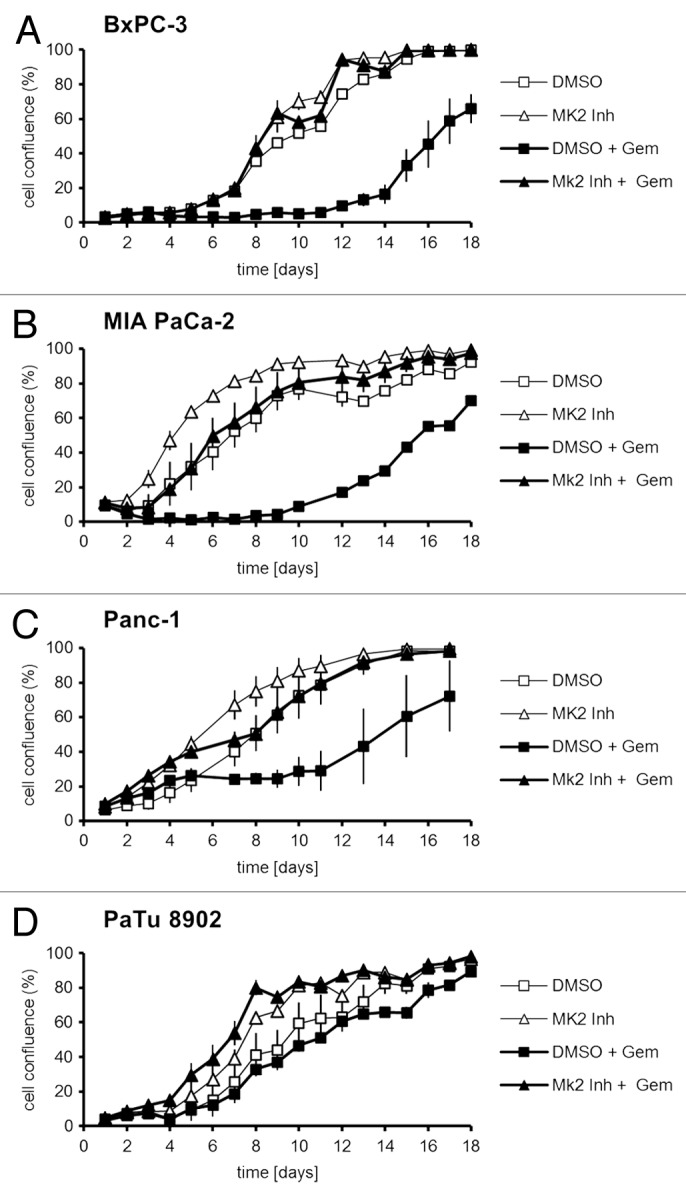
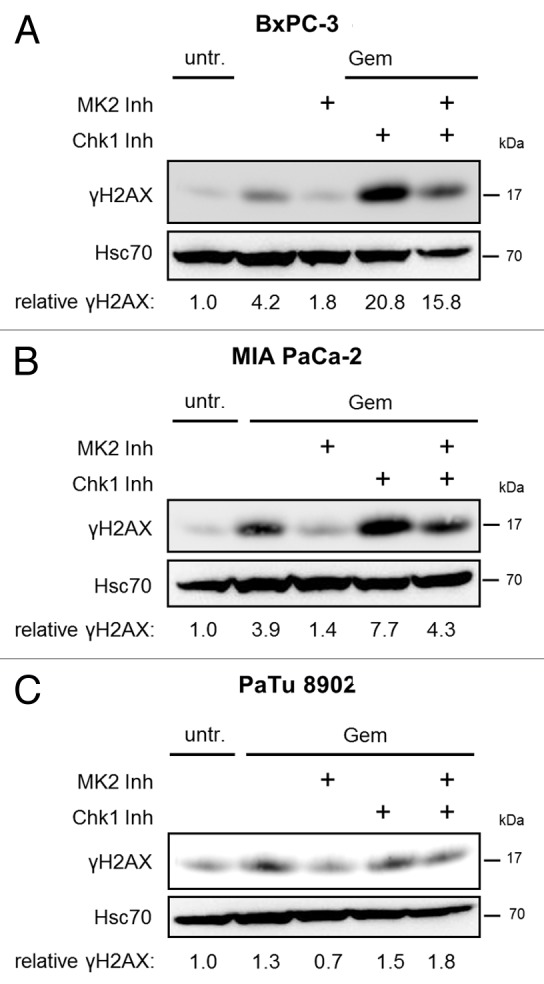
Pancreatic carcinoma is the major clinical entity where the nucleoside analog gemcitabine is used for first-line therapy. Overcoming cellular resistance toward gemcitabine remains a major challenge in this context. This raises the need to identify factors that determine gemcitabine sensitivity in pancreatic carcinoma cells. We previously found the MAPK-activated protein kinase 2 (MK2), part of the p38/MK2 stress response pathway, to be required for DNA replication fork stalling when osteosarcoma-derived cells were treated with gemcitabine. As a consequence, inhibition or depletion of MK2 protects these cells from gemcitabine-induced death (Köpper, et al. Proc Natl Acad Sci USA 2013; 110:16856-61). Here, we addressed whether MK2 also determines the sensitivity of pancreatic cancer cells toward gemcitabine. We found that MK2 inhibition reduced the intensity of the DNA damage response and enhanced survival of the pancreatic cancer cell lines BxPC-3, MIA PaCa-2, and Panc-1, which display a moderate to strong sensitivity to gemcitabine. In contrast, MK2 inhibition only weakly attenuated the DNA damage response intensity and did not enhance long-term survival in the gemcitabine-resistant cell line PaTu 8902. Importantly, in BxPC-3 and MIA PaCa-2 cells, inhibition of MK2 also rescued increased H2AX phosphorylation caused by inhibition of the checkpoint kinase Chk1 in the presence of gemcitabine. These results indicate that MK2 mediates gemcitabine efficacy in pancreatic cancer cells that respond to the drug, suggesting that the p38/MK2 pathway represents a determinant of the efficacy by that gemcitabine counteracts pancreatic cancer.
Keywords: Chk1; DNA damage; MAPKAPK2; MK2; chemotherapy; gemcitabine; pancreatic carcinoma.
Figures
Similar articles
-
Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity.Proc Natl Acad Sci U S A. 2013 Oct 15;110(42):16856-61. doi: 10.1073/pnas.1304355110. Epub 2013 Sep 30. Proc Natl Acad Sci U S A. 2013. PMID: 24082115 Free PMC article.
-
Inhibition of MAPKAPK2/MK2 facilitates DNA replication upon cancer cell treatment with gemcitabine but not cisplatin.Cancer Lett. 2018 Aug 1;428:45-54. doi: 10.1016/j.canlet.2018.04.030. Epub 2018 Apr 25. Cancer Lett. 2018. PMID: 29704518
-
Gemcitabine triggers a pro-survival response in pancreatic cancer cells through activation of the MNK2/eIF4E pathway.Oncogene. 2013 Jun 6;32(23):2848-57. doi: 10.1038/onc.2012.306. Epub 2012 Jul 16. Oncogene. 2013. PMID: 22797067
-
The Clinical Significance of Phosphorylated Heat Shock Protein 27 (HSPB1) in Pancreatic Cancer.Int J Mol Sci. 2016 Jan 21;17(1):137. doi: 10.3390/ijms17010137. Int J Mol Sci. 2016. PMID: 26805817 Free PMC article. Review.
-
Gemcitabine chemoresistance in pancreatic cancer: molecular mechanisms and potential solutions.Scand J Gastroenterol. 2009;44(7):782-6. doi: 10.1080/00365520902745039. Scand J Gastroenterol. 2009. PMID: 19214867 Review.
Cited by
-
Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells.Int J Mol Sci. 2018 Jun 6;19(6):1690. doi: 10.3390/ijms19061690. Int J Mol Sci. 2018. PMID: 29882812 Free PMC article. Review.
-
Small molecule targeting of the p38/Mk2 stress signaling pathways to improve cancer treatment.BMC Cancer. 2023 Sep 23;23(1):895. doi: 10.1186/s12885-023-11319-x. BMC Cancer. 2023. PMID: 37740222 Free PMC article.
-
Genome-scale analysis to identify prognostic microRNA biomarkers in patients with early stage pancreatic ductal adenocarcinoma after pancreaticoduodenectomy.Cancer Manag Res. 2018 Aug 10;10:2537-2551. doi: 10.2147/CMAR.S168351. eCollection 2018. Cancer Manag Res. 2018. PMID: 30127641 Free PMC article.
-
MAPK-activated protein kinase 2 is associated with poor prognosis of glioma patients and immune inhibition in glioma.Front Oncol. 2024 Jan 23;14:1307992. doi: 10.3389/fonc.2024.1307992. eCollection 2024. Front Oncol. 2024. PMID: 38322416 Free PMC article.
-
Exploiting replicative stress to treat cancer.Nat Rev Drug Discov. 2015 Jun;14(6):405-23. doi: 10.1038/nrd4553. Epub 2015 May 8. Nat Rev Drug Discov. 2015. PMID: 25953507 Review.
References
-
- Oettle H, Post S, Neuhaus P, Gellert K, Langrehr J, Ridwelski K, Schramm H, Fahlke J, Zuelke C, Burkart C, et al. Adjuvant chemotherapy with gemcitabine vs observation in patients undergoing curative-intent resection of pancreatic cancer: a randomized controlled trial. JAMA. 2007;297:267–77. doi: 10.1001/jama.297.3.267. - DOI - PubMed
-
- Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–13. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous