

Whole-genome haplotyping using long reads and statistical methods
- PMID: 24561555
- PMCID: PMC4073643
- DOI: 10.1038/nbt.2833
Whole-genome haplotyping using long reads and statistical methods
Abstract
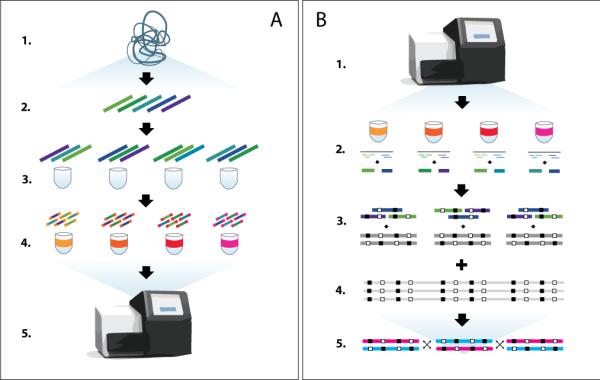
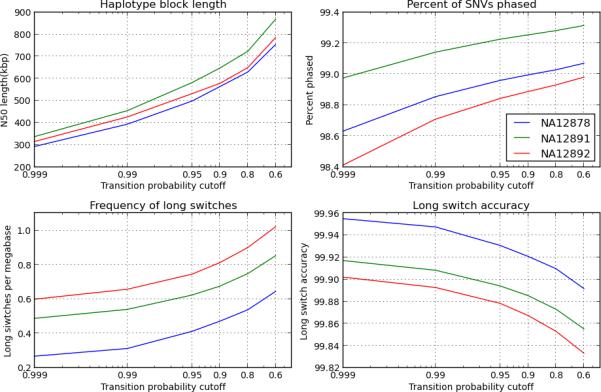
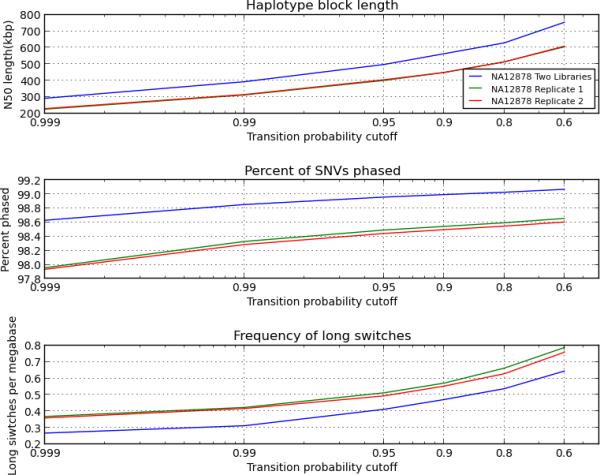
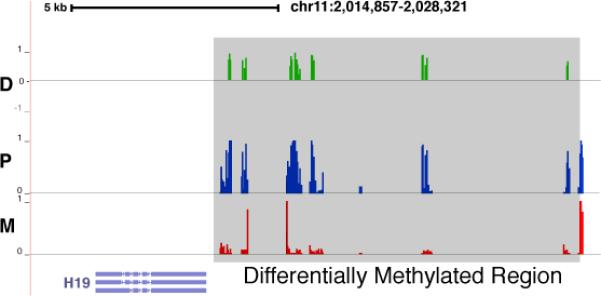
The rapid growth of sequencing technologies has greatly contributed to our understanding of human genetics. Yet, despite this growth, mainstream technologies have not been fully able to resolve the diploid nature of the human genome. Here we describe statistically aided, long-read haplotyping (SLRH), a rapid, accurate method that uses a statistical algorithm to take advantage of the partially phased information contained in long genomic fragments analyzed by short-read sequencing. For a human sample, as little as 30 Gbp of additional sequencing data are needed to phase genotypes identified by 50× coverage whole-genome sequencing. Using SLRH, we phase 99% of single-nucleotide variants in three human genomes into long haplotype blocks 0.2-1 Mbp in length. We apply our method to determine allele-specific methylation patterns in a human genome and identify hundreds of differentially methylated regions that were previously unknown. SLRH should facilitate population-scale haplotyping of human genomes.
Figures
References
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
