

Improved normalization of systematic biases affecting ion current measurements in label-free proteomics data
- PMID: 24563535
- PMCID: PMC4014289
- DOI: 10.1074/mcp.M113.030593
Improved normalization of systematic biases affecting ion current measurements in label-free proteomics data
Abstract
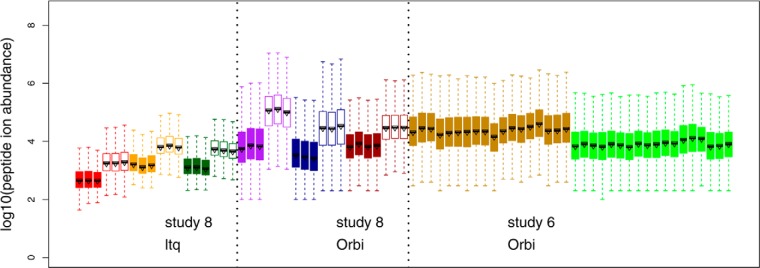
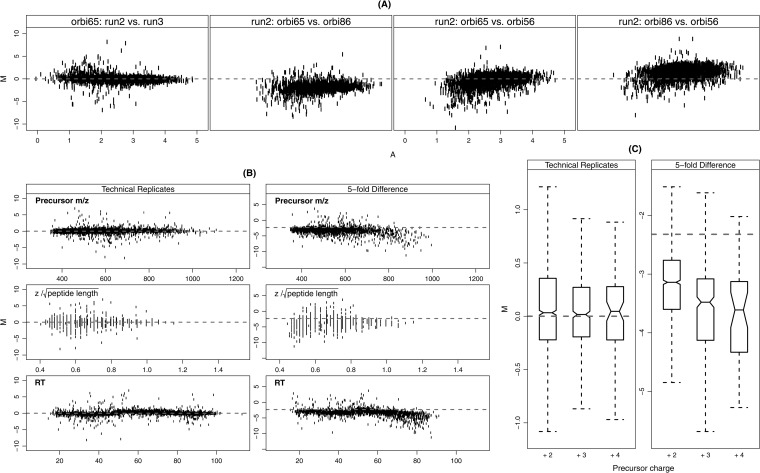
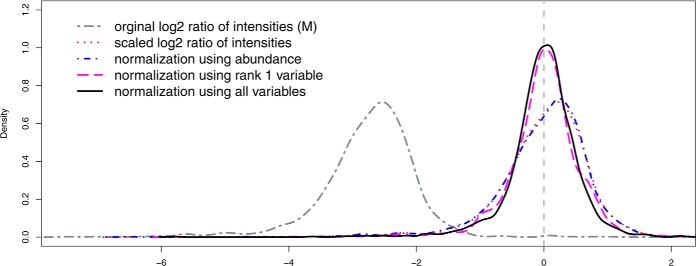
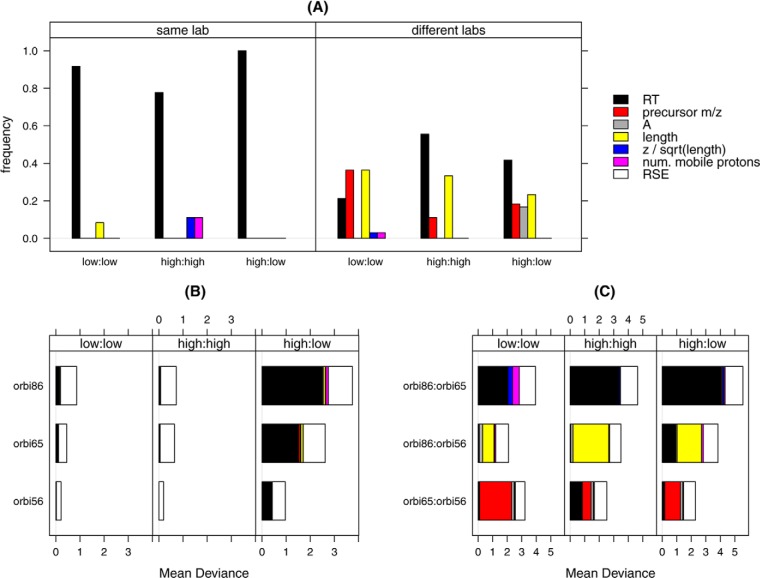
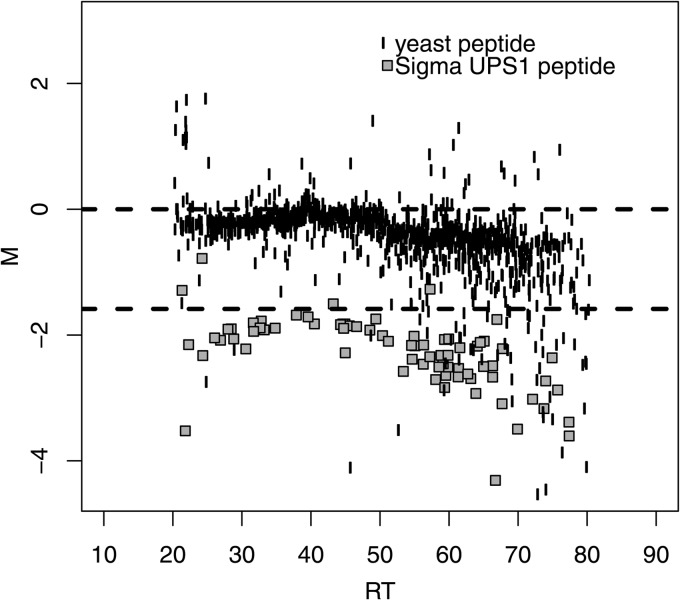
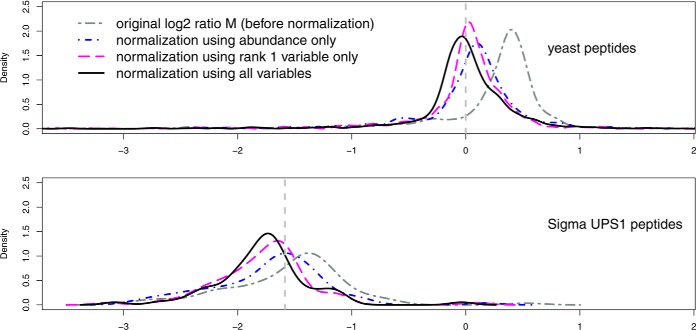
Normalization is an important step in the analysis of quantitative proteomics data. If this step is ignored, systematic biases can lead to incorrect assumptions about regulation. Most statistical procedures for normalizing proteomics data have been borrowed from genomics where their development has focused on the removal of so-called 'batch effects.' In general, a typical normalization step in proteomics works under the assumption that most peptides/proteins do not change; scaling is then used to give a median log-ratio of 0. The focus of this work was to identify other factors, derived from knowledge of the variables in proteomics, which might be used to improve normalization. Here we have examined the multi-laboratory data sets from Phase I of the NCI's CPTAC program. Surprisingly, the most important bias variables affecting peptide intensities within labs were retention time and charge state. The magnitude of these observations was exaggerated in samples of unequal concentrations or "spike-in" levels, presumably because the average precursor charge for peptides with higher charge state potentials is lower at higher relative sample concentrations. These effects are consistent with reduced protonation during electrospray and demonstrate that the physical properties of the peptides themselves can serve as good reporters of systematic biases. Between labs, retention time, precursor m/z, and peptide length were most commonly the top-ranked bias variables, over the standardly used average intensity (A). A larger set of variables was then used to develop a stepwise normalization procedure. This statistical model was found to perform as well or better on the CPTAC mock biomarker data than other commonly used methods. Furthermore, the method described here does not require a priori knowledge of the systematic biases in a given data set. These improvements can be attributed to the inclusion of variables other than average intensity during normalization.
Figures
References
-
- Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 - PubMed
-
- Mueller L. N., Brusniak M. Y., Mani D. R., Aebersold R. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res 7, 51–61 - PubMed
-
- Silva J. C., Gorenstein M. V., Li G.-Z., Vissers J. P. C., Geromanos S. J. (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell. Proteomics 5, 144–156 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
