

Microtubule defects & Neurodegeneration
- PMID: 24563812
- PMCID: PMC3930179
- DOI: 10.4172/2157-7412.1000203
Microtubule defects & Neurodegeneration
Abstract
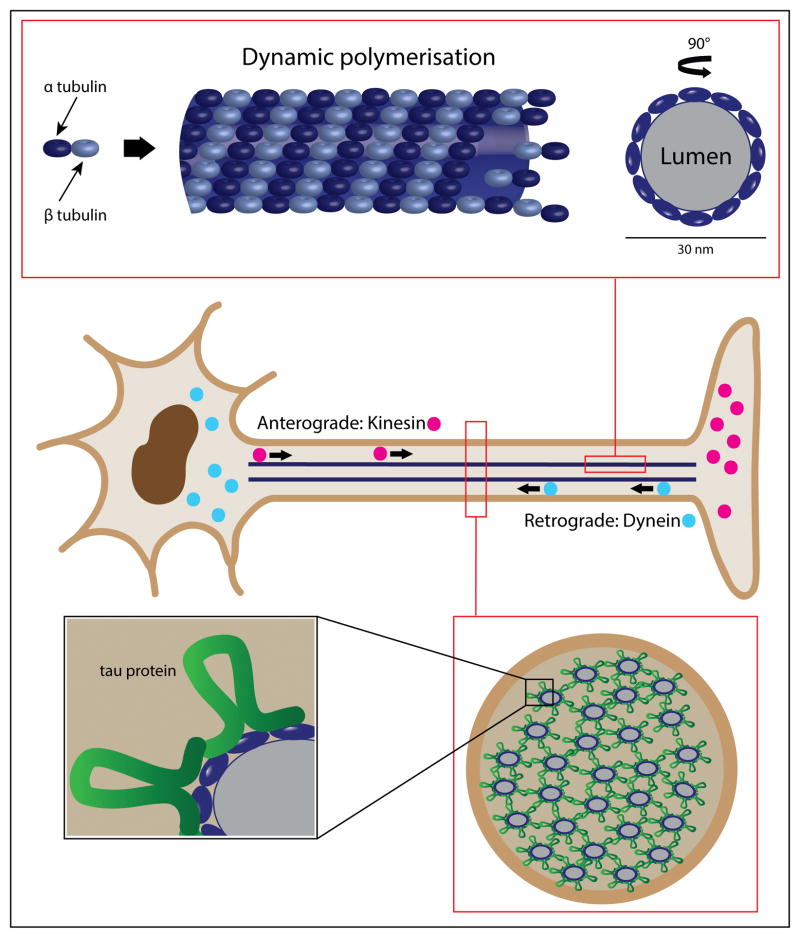
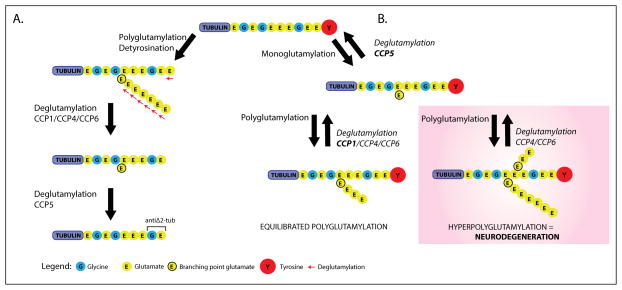
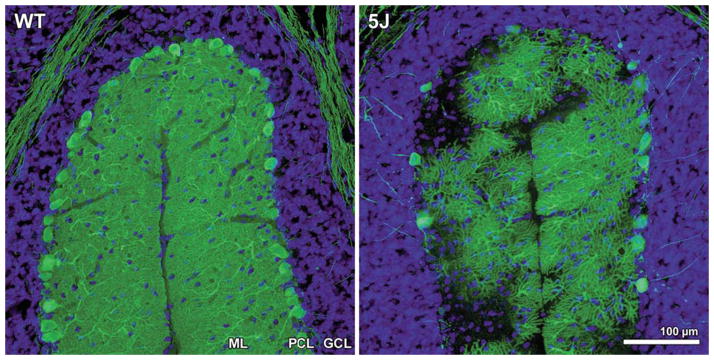
One of the major challenges facing the long term survival of neurons is their requirement to maintain efficient axonal transport over long distances. In humans as large, long-lived vertebrates, the machinery maintaining neuronal transport must remain efficient despite the slow accumulation of cell damage during aging. Mutations in genes encoding proteins which function in the transport system feature prominently in neurologic disorders. Genes known to cause such disorders and showing traditional Mendelian inheritance have been more readily identified. It has been more difficult, however, to isolate factors underlying the complex genetics contributing to the more common idiopathic forms of neurodegenerative disease. At the heart of neuronal transport is the rail network or scaffolding provided by neuron specific microtubules (MTs). The importance of MT dynamics and stability is underscored by the critical role tau protein plays in MT-associated stabilization versus the dysfunction seen in Alzheimer's disease, frontotemporal dementia and other tauopathies. Another example of the requirement for tight regulation of MT dynamics is the need to maintain balanced levels of post-translational modification of key MT building-blocks such as α-tubulin. Tubulins require extensive polyglutamylation at their carboxyl-terminus as part of a novel post-translational modification mechanism to signal MT growth versus destabilization. Dramatically, knock-out of a gene encoding a deglutamylation family member causes an extremely rapid cell death of Purkinje cells in the ataxic mouse model, pcd. This review will examine a range of neurodegenerative conditions where current molecular understanding points to defects in the stability of MTs and axonal transport to emphasize the central role of MTs in neuron survival.
Figures
References
-
- Audebert S, Koulakoff A, Berwald-Netter Y, Gros F, Denoulet P, Edde B. Developmental regulation of polyglutamylated alpha- and beta-tubulin in mouse brain neurons. J Cell Sci. 1994;107 (Pt 8):2313–2322. - PubMed
-
- Barra HS, Arce CA, Argarana CE. Posttranslational tyrosination/detyrosination of tubulin. Mol Neurobiol. 1988;2 (2):133–153. - PubMed
-
- Ben Othmane K, Middleton LT, Loprest LJ, Wilkinson KM, Lennon F, Rozear MP, Stajich JM, Gaskell PC, Roses AD, Pericak-Vance MA, et al. Localization of a gene (CMT2A) for autosomal dominant Charcot-Marie-Tooth disease type 2 to chromosome 1p and evidence of genetic heterogeneity. Genomics. 1993;17 (2):370–375. - PubMed
-
- Center for Drug Evaluation R, & Center for Biologics Evaluation and Research. Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics. F. a. D. A. U. S. A. U.S. Department of Health and Human Services; 2013.
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources