

Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives
- PMID: 24564169
- PMCID: PMC3846878
- DOI: 10.1186/1471-2105-14-S11-S1
Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives
Abstract
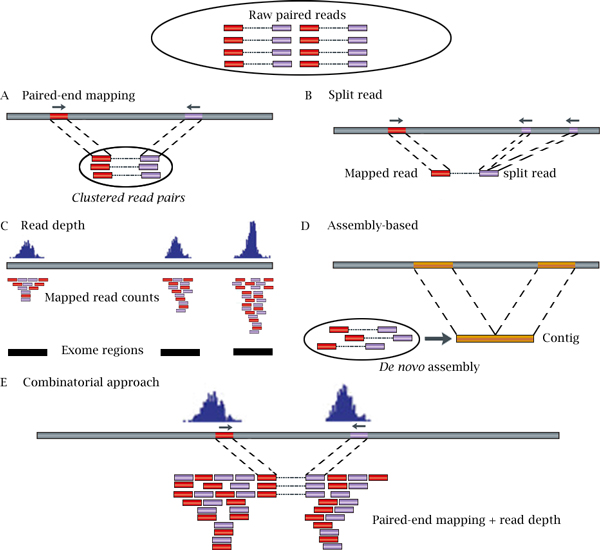
Copy number variation (CNV) is a prevalent form of critical genetic variation that leads to an abnormal number of copies of large genomic regions in a cell. Microarray-based comparative genome hybridization (arrayCGH) or genotyping arrays have been standard technologies to detect large regions subject to copy number changes in genomes until most recently high-resolution sequence data can be analyzed by next-generation sequencing (NGS). During the last several years, NGS-based analysis has been widely applied to identify CNVs in both healthy and diseased individuals. Correspondingly, the strong demand for NGS-based CNV analyses has fuelled development of numerous computational methods and tools for CNV detection. In this article, we review the recent advances in computational methods pertaining to CNV detection using whole genome and whole exome sequencing data. Additionally, we discuss their strengths and weaknesses and suggest directions for future development.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
