

MetaCluster-TA: taxonomic annotation for metagenomic data based on assembly-assisted binning
- PMID: 24564377
- PMCID: PMC4046714
- DOI: 10.1186/1471-2164-15-S1-S12
MetaCluster-TA: taxonomic annotation for metagenomic data based on assembly-assisted binning
Abstract
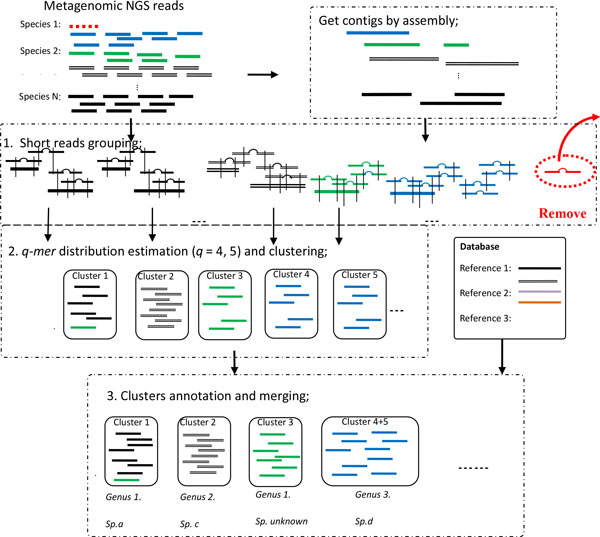
Background: Taxonomic annotation of reads is an important problem in metagenomic analysis. Existing annotation tools, which rely on the approach of aligning each read to the taxonomic structure, are unable to annotate many reads efficiently and accurately as reads (~100 bp) are short and most of them come from unknown genomes. Previous work has suggested assembling the reads to make longer contigs before annotation. More reads/contigs can be annotated as a longer contig (in Kbp) can be aligned to a taxon even if it is from an unknown species as long as it contains a conserved region of that taxon. Unfortunately existing metagenomic assembly tools are not mature enough to produce long enough contigs. Binning tries to group reads/contigs of similar species together. Intuitively, reads in the same group (cluster) should be annotated to the same taxon and these reads altogether should cover a significant portion of the genome alleviating the problem of short contigs if the quality of binning is high. However, no existing work has tried to use binning results to help solve the annotation problem. This work explores this direction.
Results: In this paper, we describe MetaCluster-TA, an assembly-assisted binning-based annotation tool which relies on an innovative idea of annotating binned reads instead of aligning each read or contig to the taxonomic structure separately. We propose the novel concept of the 'virtual contig' (which can be up to 10 Kb in length) to represent a set of reads and then represent each cluster as a set of 'virtual contigs' (which together can be total up to 1 Mb in length) for annotation. MetaCluster-TA can outperform widely-used MEGAN4 and can annotate (1) more reads since the virtual contigs are much longer; (2) more accurately since each cluster of long virtual contigs contains global information of the sampled genome which tends to be more accurate than short reads or assembled contigs which contain only local information of the genome; and (3) more efficiently since there are much fewer long virtual contigs to align than short reads. MetaCluster-TA outperforms MetaCluster 5.0 as a binning tool since binning itself can be more sensitive and precise given long virtual contigs and the binning results can be improved using the reference taxonomic database.
Conclusions: MetaCluster-TA can outperform widely-used MEGAN4 and can annotate more reads with higher accuracy and higher efficiency. It also outperforms MetaCluster 5.0 as a binning tool.
Similar articles
-
Exploiting topic modeling to boost metagenomic reads binning.BMC Bioinformatics. 2015;16 Suppl 5(Suppl 5):S2. doi: 10.1186/1471-2105-16-S5-S2. Epub 2015 Mar 18. BMC Bioinformatics. 2015. PMID: 25859745 Free PMC article.
-
Metagenome Assembly and Contig Assignment.Methods Mol Biol. 2018;1849:179-192. doi: 10.1007/978-1-4939-8728-3_12. Methods Mol Biol. 2018. PMID: 30298255
-
METAMVGL: a multi-view graph-based metagenomic contig binning algorithm by integrating assembly and paired-end graphs.BMC Bioinformatics. 2021 Jul 22;22(Suppl 10):378. doi: 10.1186/s12859-021-04284-4. BMC Bioinformatics. 2021. PMID: 34294039 Free PMC article.
-
Genome-resolved metagenomics using environmental and clinical samples.Brief Bioinform. 2021 Sep 2;22(5):bbab030. doi: 10.1093/bib/bbab030. Brief Bioinform. 2021. PMID: 33758906 Free PMC article. Review.
-
Genome-resolved metagenomics from short-read sequencing data in the era of artificial intelligence.Funct Integr Genomics. 2025 Jun 10;25(1):124. doi: 10.1007/s10142-025-01625-x. Funct Integr Genomics. 2025. PMID: 40493087 Review.
Cited by
-
The Road to Metagenomics: From Microbiology to DNA Sequencing Technologies and Bioinformatics.Front Genet. 2015 Dec 17;6:348. doi: 10.3389/fgene.2015.00348. eCollection 2015. Front Genet. 2015. PMID: 26734060 Free PMC article. Review.
-
High-resolution characterization of the human microbiome.Transl Res. 2017 Jan;179:7-23. doi: 10.1016/j.trsl.2016.07.012. Epub 2016 Jul 25. Transl Res. 2017. PMID: 27513210 Free PMC article. Review.
-
Contrasting modes of mitochondrial genome evolution in sister taxa of wood-eating marine bivalves (Teredinidae and Xylophagaidae).Genome Biol Evol. 2022 Jun 17;14(6):evac089. doi: 10.1093/gbe/evac089. Online ahead of print. Genome Biol Evol. 2022. PMID: 35714221 Free PMC article.
-
Bioinformatics strategies for taxonomy independent binning and visualization of sequences in shotgun metagenomics.Comput Struct Biotechnol J. 2016 Dec 5;15:48-55. doi: 10.1016/j.csbj.2016.11.005. eCollection 2017. Comput Struct Biotechnol J. 2016. PMID: 27980708 Free PMC article. Review.
-
A novel semi-supervised algorithm for the taxonomic assignment of metagenomic reads.BMC Bioinformatics. 2016 Jan 6;17:22. doi: 10.1186/s12859-015-0872-x. BMC Bioinformatics. 2016. PMID: 26740458 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
