

MetaID: a novel method for identification and quantification of metagenomic samples
- PMID: 24564518
- PMCID: PMC4042266
- DOI: 10.1186/1471-2164-14-S8-S4
MetaID: a novel method for identification and quantification of metagenomic samples
Abstract
Background: Advances in next-generation sequencing (NGS) technology has provided us with an opportunity to analyze and evaluate the rich microbial communities present in all natural environments. The shorter reads obtained from the shortgun technology has paved the way for determining the taxonomic profile of a community by simply aligning the reads against the available reference genomes. While several computational methods are available for taxonomic profiling at the genus- and species-level, none of these methods are effective at the strain-level identification due to the increasing difficulty in detecting variation at that level. Here, we present MetaID, an alignment-free n-gram based approach that can accurately identify microorganisms at the strain level and estimate the abundance of each organism in a sample, given a metagenomic sequencing dataset.
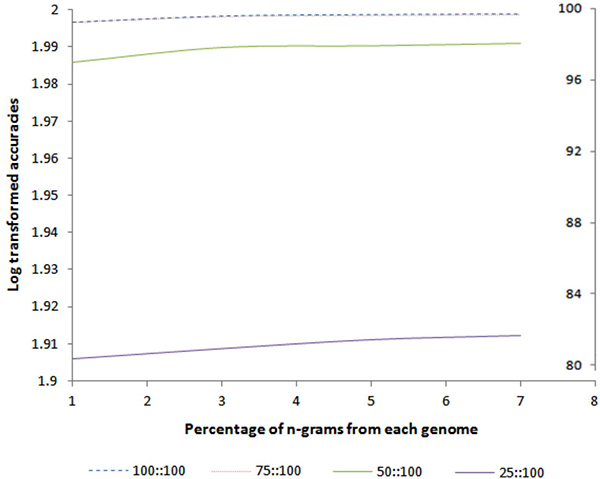
Results: MetaID is an n-gram based method that calculates the profile of unique and common n-grams from the dataset of 2,031 prokaryotic genomes and assigns weights to each n-gram using a scoring function. This scoring function assigns higher weightage to the n-grams that appear in fewer genomes and vice versa; thus, allows for effective use of both unique and common n-grams for species identification. Our 10-fold cross-validation results on a simulated dataset show a remarkable accuracy of 99.7% at the strain-level identification of the organisms in gut microbiome. We also demonstrated that our model shows impressive performance even by using only 25% or 50% of the genome sequences for modeling. In addition to identification of the species, our method can also estimate the relative abundance of each species in the simulated metagenomic samples. The generic approach employed in this method can be applied for accurate identification of a wide variety of microbial species (viruses, prokaryotes and eukaryotes) present in any environmental sample.
Conclusions: The proposed scoring function and approach is able to accurately identify and estimate the entire taxa in any metagenomic community. The weights assigned to the common n-grams by our scoring function are precisely calibrated to match the reads up to the strain level. Our multipronged validation tests demonstrate that MetaID is sufficiently robust to accurately identify and estimate the abundance of each taxon in any natural environment even when using incomplete or partially sequenced genomes.
Figures




Similar articles
-
StrainIQ: A Novel n-Gram-Based Method for Taxonomic Profiling of Human Microbiota at the Strain Level.Genes (Basel). 2023 Aug 18;14(8):1647. doi: 10.3390/genes14081647. Genes (Basel). 2023. PMID: 37628698 Free PMC article.
-
Human reference gut microbiome catalog including newly assembled genomes from under-represented Asian metagenomes.Genome Med. 2021 Aug 27;13(1):134. doi: 10.1186/s13073-021-00950-7. Genome Med. 2021. PMID: 34446072 Free PMC article.
-
A statistical framework for accurate taxonomic assignment of metagenomic sequencing reads.PLoS One. 2012;7(10):e46450. doi: 10.1371/journal.pone.0046450. Epub 2012 Oct 1. PLoS One. 2012. PMID: 23049702 Free PMC article.
-
Assessment of metagenomic assemblers based on hybrid reads of real and simulated metagenomic sequences.Brief Bioinform. 2020 May 21;21(3):777-790. doi: 10.1093/bib/bbz025. Brief Bioinform. 2020. PMID: 30860572 Free PMC article. Review.
-
Solving genomic puzzles: computational methods for metagenomic binning.Brief Bioinform. 2024 Jul 25;25(5):bbae372. doi: 10.1093/bib/bbae372. Brief Bioinform. 2024. PMID: 39082646 Free PMC article. Review.
Cited by
-
WGSQuikr: fast whole-genome shotgun metagenomic classification.PLoS One. 2014 Mar 13;9(3):e91784. doi: 10.1371/journal.pone.0091784. eCollection 2014. PLoS One. 2014. PMID: 24626336 Free PMC article.
-
StrainIQ: A Novel n-Gram-Based Method for Taxonomic Profiling of Human Microbiota at the Strain Level.Genes (Basel). 2023 Aug 18;14(8):1647. doi: 10.3390/genes14081647. Genes (Basel). 2023. PMID: 37628698 Free PMC article.
-
Considerations for Optimization of High-Throughput Sequencing Bioinformatics Pipelines for Virus Detection.Viruses. 2018 Sep 27;10(10):528. doi: 10.3390/v10100528. Viruses. 2018. PMID: 30262776 Free PMC article.
-
Detection of somatic mutations in tumors using unaligned clonal sequencing data.Lab Invest. 2014 Oct;94(10):1173-83. doi: 10.1038/labinvest.2014.96. Epub 2014 Jul 28. Lab Invest. 2014. PMID: 25068661
-
Interdisciplinary dialogue for education, collaboration, and innovation: intelligent Biology and Medicine in and beyond 2013.BMC Genomics. 2013;14 Suppl 8(Suppl 8):S1. doi: 10.1186/1471-2164-14-S8-S1. Epub 2013 Dec 9. BMC Genomics. 2013. PMID: 24564388 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
