

Using distances between Top-n-gram and residue pairs for protein remote homology detection
- PMID: 24564580
- PMCID: PMC4015815
- DOI: 10.1186/1471-2105-15-S2-S3
Using distances between Top-n-gram and residue pairs for protein remote homology detection
Abstract
Background: Protein remote homology detection is one of the central problems in bioinformatics, which is important for both basic research and practical application. Currently, discriminative methods based on Support Vector Machines (SVMs) achieve the state-of-the-art performance. Exploring feature vectors incorporating the position information of amino acids or other protein building blocks is a key step to improve the performance of the SVM-based methods.
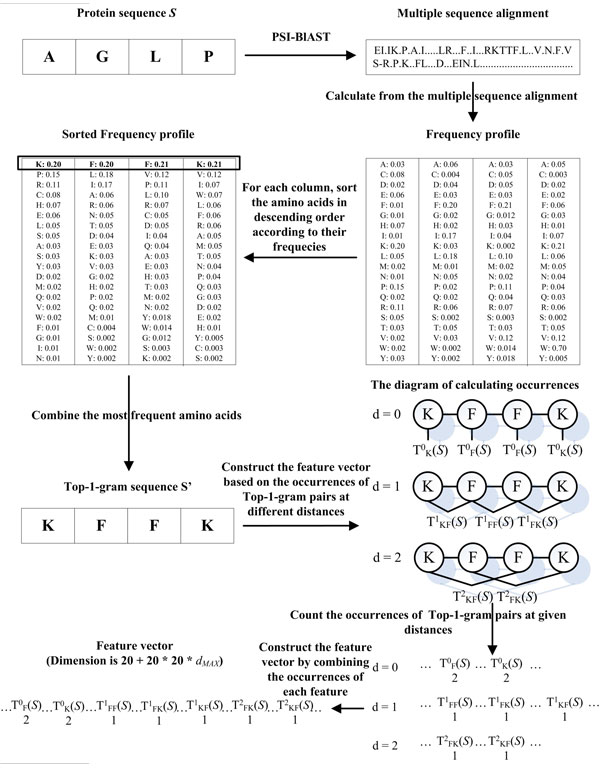
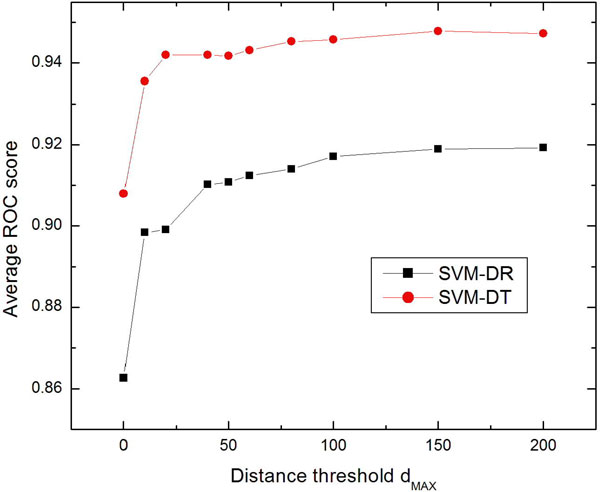
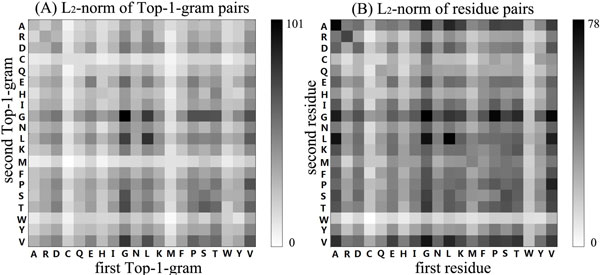
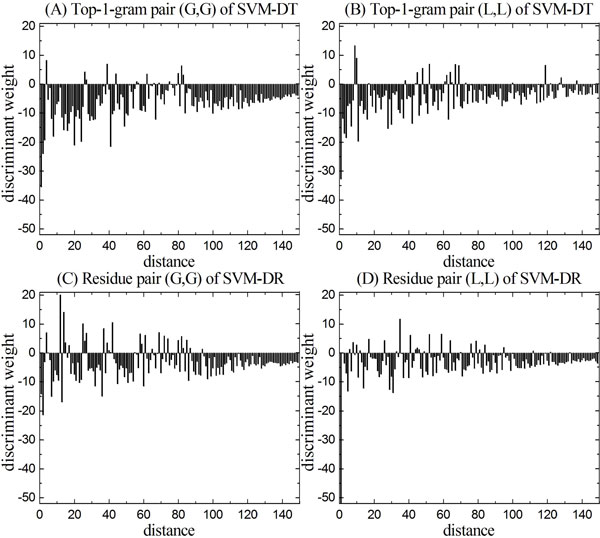
Results: Two new methods for protein remote homology detection were proposed, called SVM-DR and SVM-DT. SVM-DR is a sequence-based method, in which the feature vector representation for protein is based on the distances between residue pairs. SVM-DT is a profile-based method, which considers the distances between Top-n-gram pairs. Top-n-gram can be viewed as a profile-based building block of proteins, which is calculated from the frequency profiles. These two methods are position dependent approaches incorporating the sequence-order information of protein sequences. Various experiments were conducted on a benchmark dataset containing 54 families and 23 superfamilies. Experimental results showed that these two new methods are very promising. Compared with the position independent methods, the performance improvement is obvious. Furthermore, the proposed methods can also provide useful insights for studying the features of protein families.
Conclusion: The better performance of the proposed methods demonstrates that the position dependant approaches are efficient for protein remote homology detection. Another advantage of our methods arises from the explicit feature space representation, which can be used to analyze the characteristic features of protein families. The source code of SVM-DT and SVM-DR is available at http://bioinformatics.hitsz.edu.cn/DistanceSVM/index.jsp.
Figures

Similar articles
-
Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection.Bioinformatics. 2014 Feb 15;30(4):472-9. doi: 10.1093/bioinformatics/btt709. Epub 2013 Dec 5. Bioinformatics. 2014. PMID: 24318998 Free PMC article.
-
Using amino acid physicochemical distance transformation for fast protein remote homology detection.PLoS One. 2012;7(9):e46633. doi: 10.1371/journal.pone.0046633. Epub 2012 Sep 28. PLoS One. 2012. PMID: 23029559 Free PMC article.
-
SVM-Fold: a tool for discriminative multi-class protein fold and superfamily recognition.BMC Bioinformatics. 2007 May 22;8 Suppl 4(Suppl 4):S2. doi: 10.1186/1471-2105-8-S4-S2. BMC Bioinformatics. 2007. PMID: 17570145 Free PMC article.
-
A comprehensive review and comparison of different computational methods for protein remote homology detection.Brief Bioinform. 2018 Mar 1;19(2):231-244. doi: 10.1093/bib/bbw108. Brief Bioinform. 2018. PMID: 27881430 Review.
-
The WWWH of remote homolog detection: the state of the art.Brief Bioinform. 2007 Mar;8(2):78-87. doi: 10.1093/bib/bbl032. Epub 2006 Sep 26. Brief Bioinform. 2007. PMID: 17003074 Review.
Cited by
-
CarSite-II: an integrated classification algorithm for identifying carbonylated sites based on K-means similarity-based undersampling and synthetic minority oversampling techniques.BMC Bioinformatics. 2021 Apr 26;22(1):216. doi: 10.1186/s12859-021-04134-3. BMC Bioinformatics. 2021. PMID: 33902446 Free PMC article.
-
Protein binding site prediction by combining hidden Markov support vector machine and profile-based propensities.ScientificWorldJournal. 2014;2014:464093. doi: 10.1155/2014/464093. Epub 2014 Jul 14. ScientificWorldJournal. 2014. PMID: 25133234 Free PMC article.
-
A novel two-way rebalancing strategy for identifying carbonylation sites.BMC Bioinformatics. 2023 Nov 13;24(1):429. doi: 10.1186/s12859-023-05551-2. BMC Bioinformatics. 2023. PMID: 37957582 Free PMC article.
-
dRHP-PseRA: detecting remote homology proteins using profile-based pseudo protein sequence and rank aggregation.Sci Rep. 2016 Sep 1;6:32333. doi: 10.1038/srep32333. Sci Rep. 2016. PMID: 27581095 Free PMC article.
-
Design of Protein Segments and Peptides for Binding to Protein Targets.Biodes Res. 2022 Apr 15;2022:9783197. doi: 10.34133/2022/9783197. eCollection 2022. Biodes Res. 2022. PMID: 37850124 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
