

Disordered amyloidogenic peptides may insert into the membrane and assemble into common cyclic structural motifs
- PMID: 24566672
- PMCID: PMC4143503
- DOI: 10.1039/c3cs60459d
Disordered amyloidogenic peptides may insert into the membrane and assemble into common cyclic structural motifs
Abstract
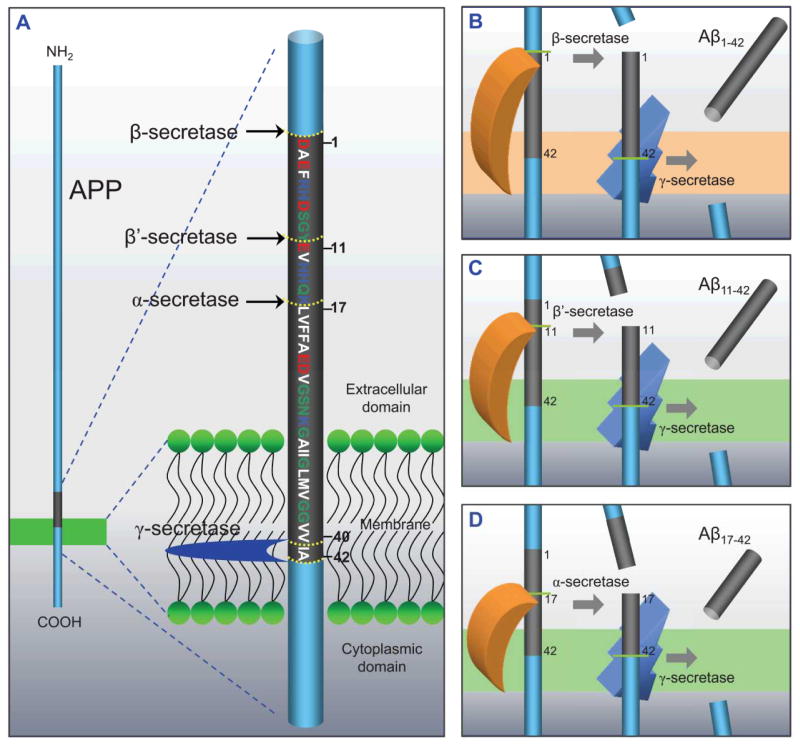
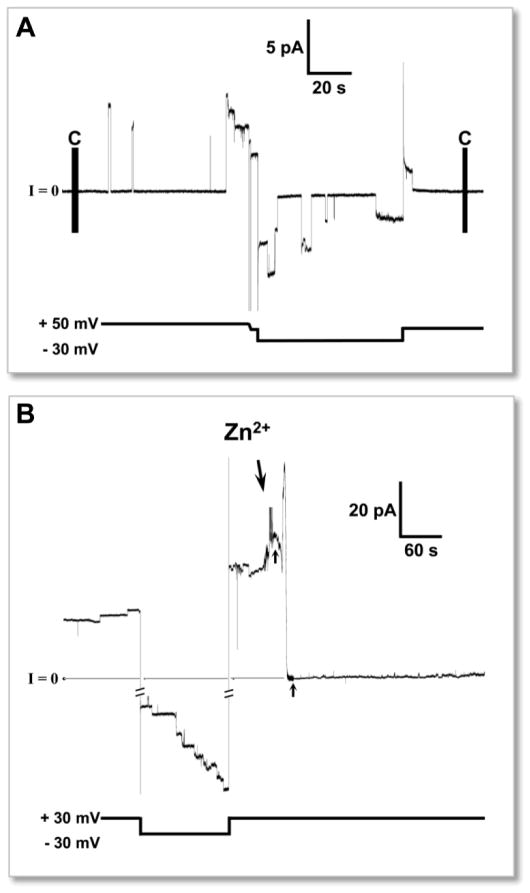
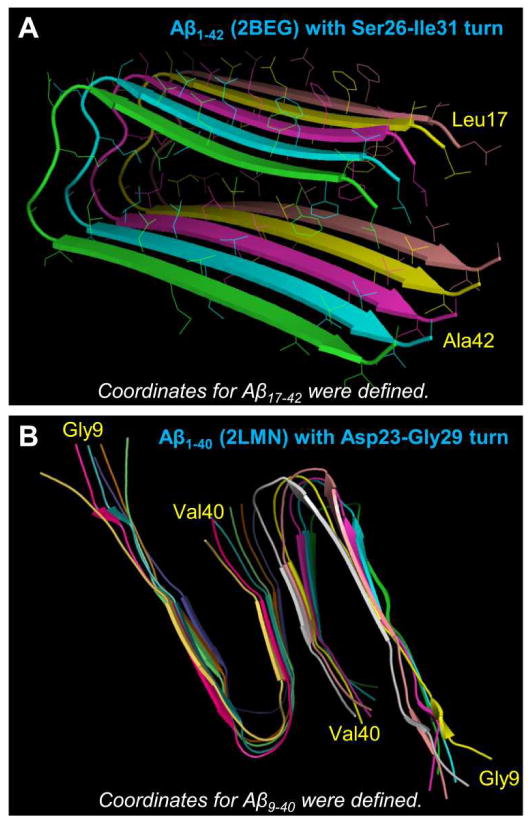
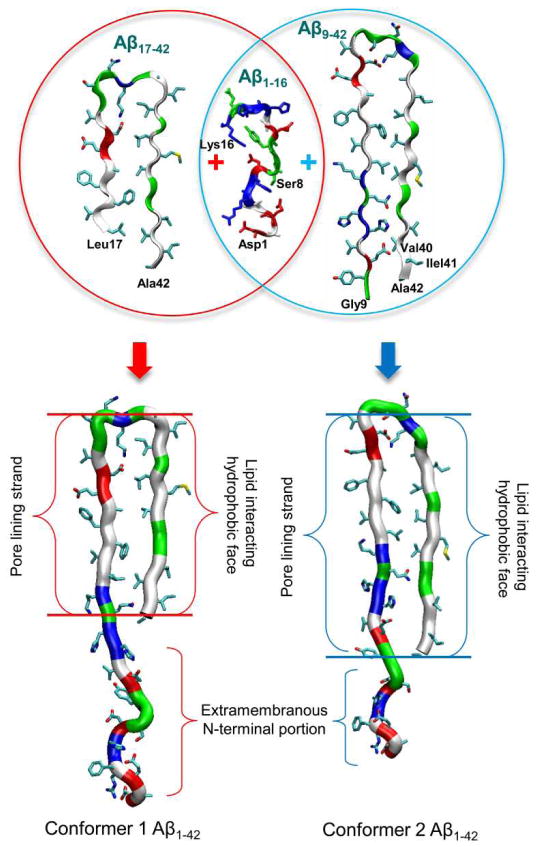
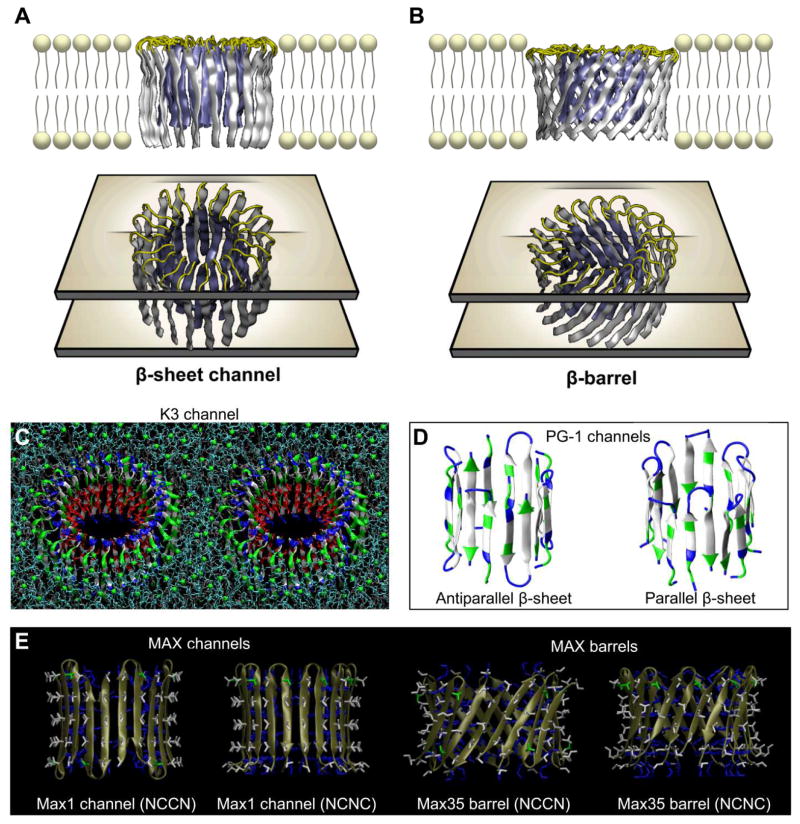
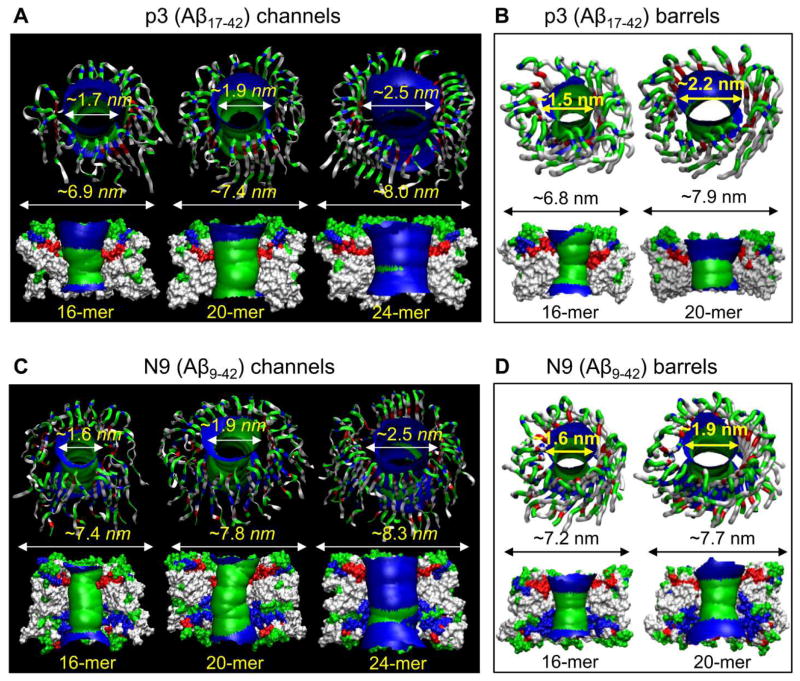
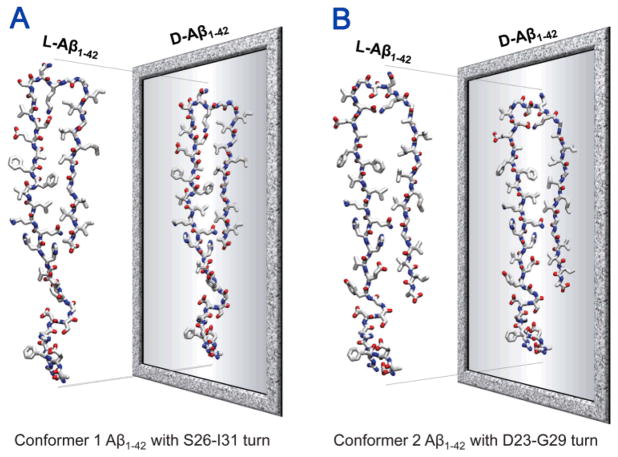
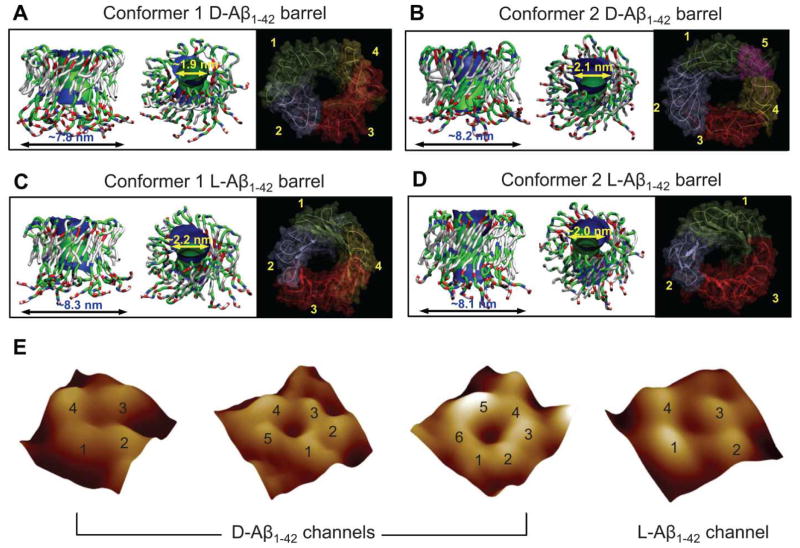
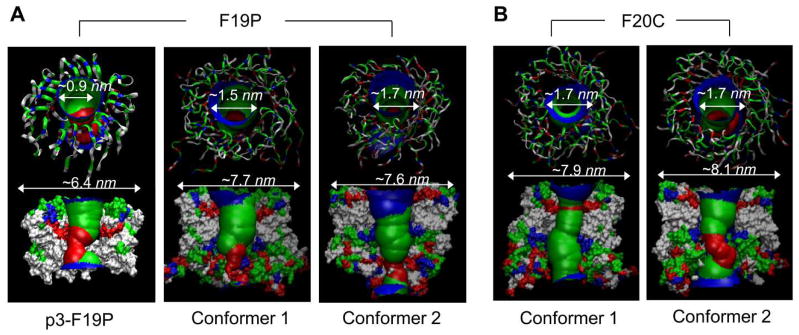
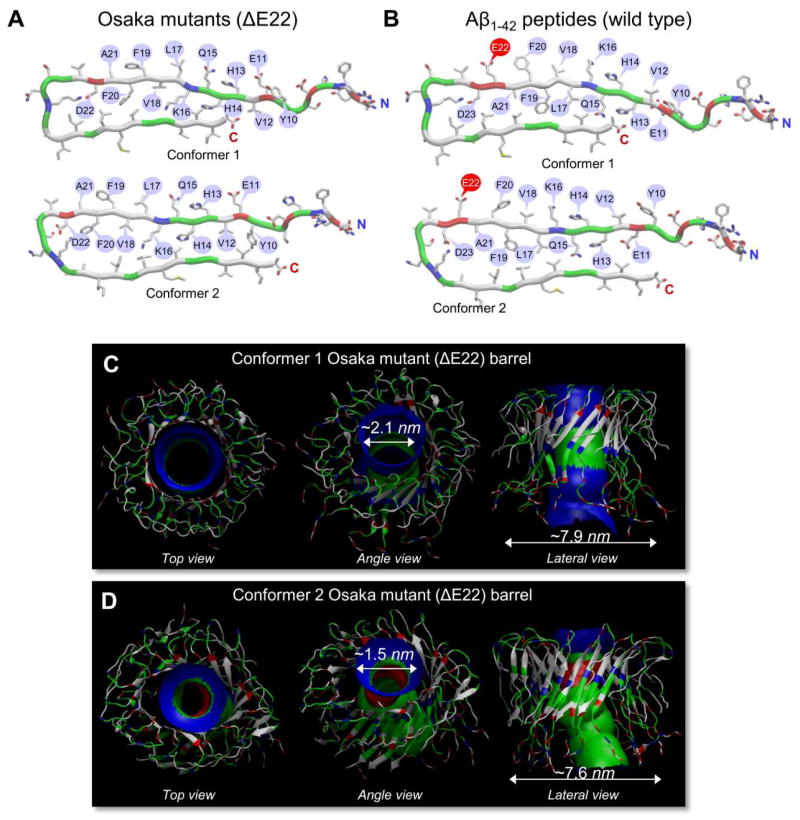
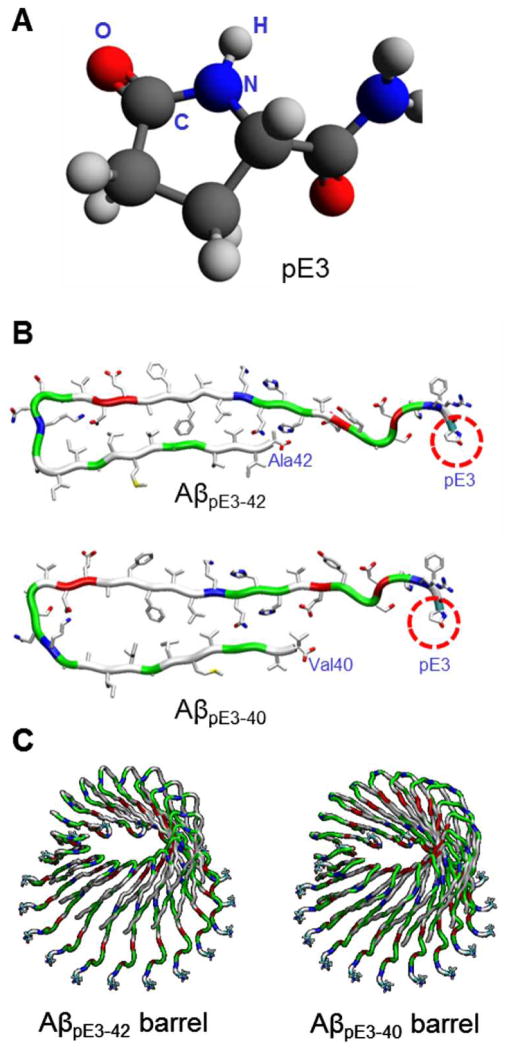
Aggregation of disordered amyloidogenic peptides into oligomers is the causative agent of amyloid-related diseases. In solution, disordered protein states are characterized by heterogeneous ensembles. Among these, β-rich conformers self-assemble via a conformational selection mechanism to form energetically-favored cross-β structures, regardless of their precise sequences. These disordered peptides can also penetrate the membrane, and electrophysiological data indicate that they form ion-conducting channels. Based on these and additional data, including imaging and molecular dynamics simulations of a range of amyloid peptides, Alzheimer's amyloid-β (Aβ) peptide, its disease-related variants with point mutations and N-terminal truncated species, other amyloidogenic peptides, as well as a cytolytic peptide and a synthetic gel-forming peptide, we suggest that disordered amyloidogenic peptides can also present a common motif in the membrane. The motif consists of curved, moon-like β-rich oligomers associated into annular organizations. The motif is favored in the lipid bilayer since it permits hydrophobic side chains to face and interact with the membrane and the charged/polar residues to face the solvated channel pores. Such channels are toxic since their pores allow uncontrolled leakage of ions into/out of the cell, destabilizing cellular ionic homeostasis. Here we detail Aβ, whose aggregation is associated with Alzheimer's disease (AD) and for which there are the most abundant data. AD is a protein misfolding disease characterized by a build-up of Aβ peptide as senile plaques, neurodegeneration, and memory loss. Excessively produced Aβ peptides may directly induce cellular toxicity, even without the involvement of membrane receptors through Aβ peptide-plasma membrane interactions.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources