

Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries
- PMID: 24568772
- PMCID: PMC3824117
- DOI: 10.1016/j.ajhg.2013.10.002
Pulling out the 1%: whole-genome capture for the targeted enrichment of ancient DNA sequencing libraries
Abstract
Most ancient specimens contain very low levels of endogenous DNA, precluding the shotgun sequencing of many interesting samples because of cost. Ancient DNA (aDNA) libraries often contain <1% endogenous DNA, with the majority of sequencing capacity taken up by environmental DNA. Here we present a capture-based method for enriching the endogenous component of aDNA sequencing libraries. By using biotinylated RNA baits transcribed from genomic DNA libraries, we are able to capture DNA fragments from across the human genome. We demonstrate this method on libraries created from four Iron Age and Bronze Age human teeth from Bulgaria, as well as bone samples from seven Peruvian mummies and a Bronze Age hair sample from Denmark. Prior to capture, shotgun sequencing of these libraries yielded an average of 1.2% of reads mapping to the human genome (including duplicates). After capture, this fraction increased substantially, with up to 59% of reads mapped to human and enrichment ranging from 6- to 159-fold. Furthermore, we maintained coverage of the majority of regions sequenced in the precapture library. Intersection with the 1000 Genomes Project reference panel yielded an average of 50,723 SNPs (range 3,062-147,243) for the postcapture libraries sequenced with 1 million reads, compared with 13,280 SNPs (range 217-73,266) for the precapture libraries, increasing resolution in population genetic analyses. Our whole-genome capture approach makes it less costly to sequence aDNA from specimens containing very low levels of endogenous DNA, enabling the analysis of larger numbers of samples.
Copyright © 2013 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
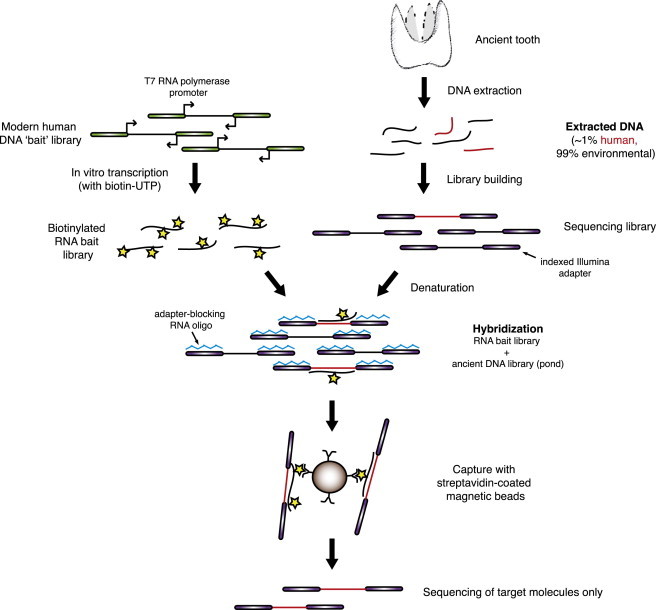
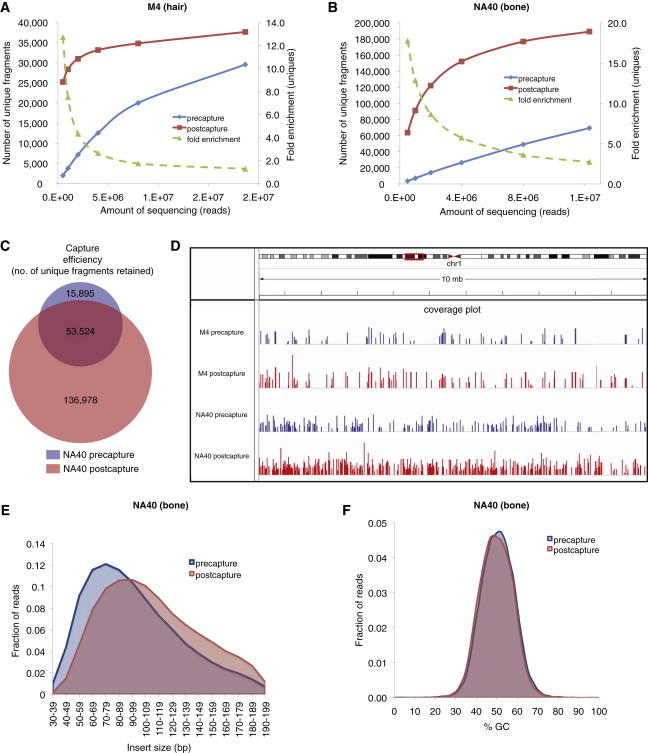
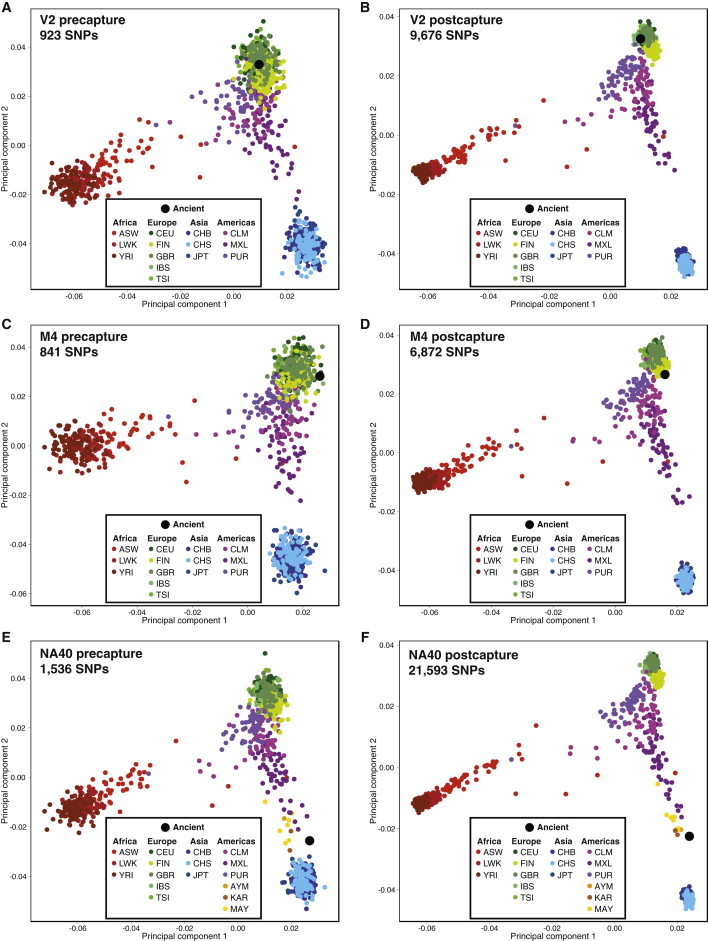
References
-
- Keller A., Graefen A., Ball M., Matzas M., Boisguerin V., Maixner F., Leidinger P., Backes C., Khairat R., Forster M. New insights into the Tyrolean Iceman’s origin and phenotype as inferred by whole-genome sequencing. Nat Commun. 2012;3:698. - PubMed
-
- Sánchez-Quinto F., Schroeder H., Ramirez O., Avila-Arcos M.C., Pybus M., Olalde I., Velazquez A.M., Marcos M.E., Encinas J.M., Bertranpetit J. Genomic affinities of two 7,000-year-old Iberian hunter-gatherers. Curr. Biol. 2012;22:1494–1499. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
