

Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma
- PMID: 24569374
- PMCID: PMC3934165
- DOI: 10.1172/JCI70454
Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma
Abstract
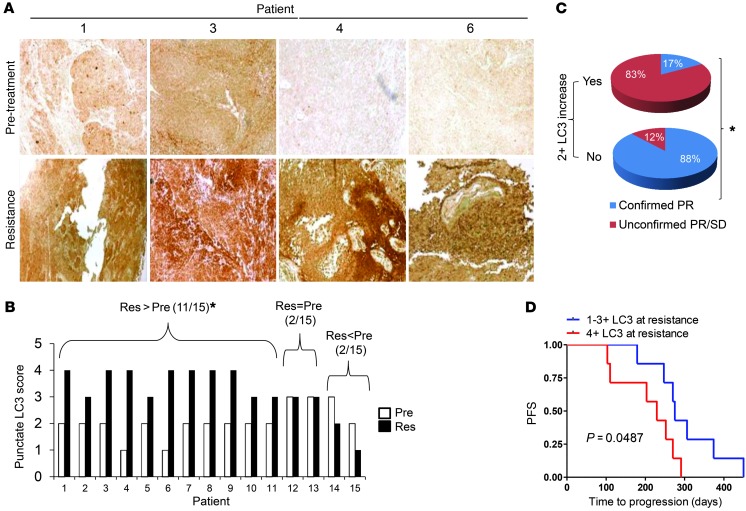
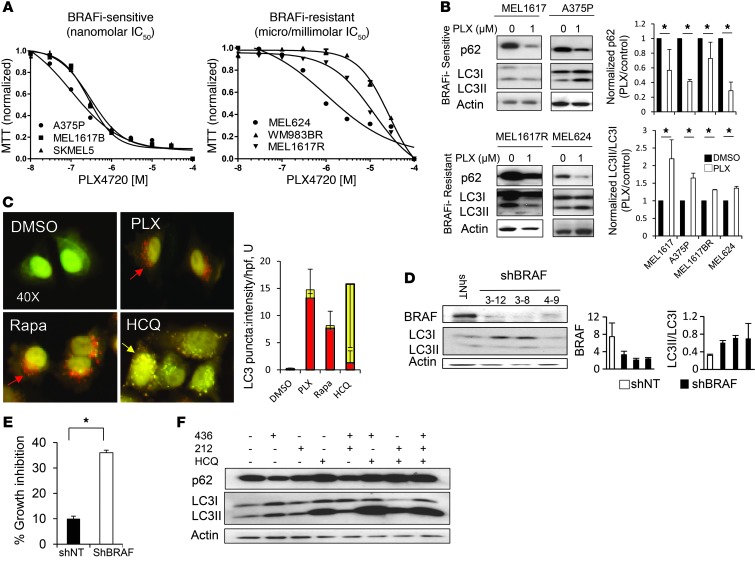
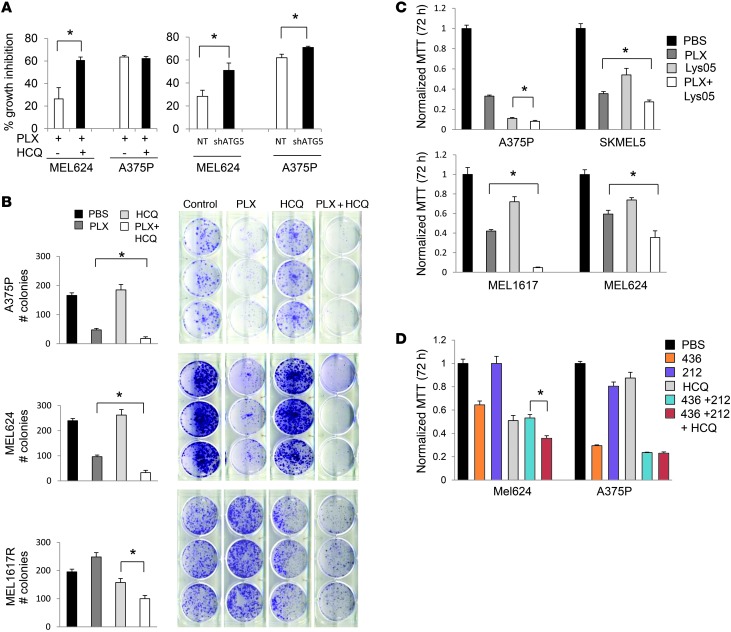
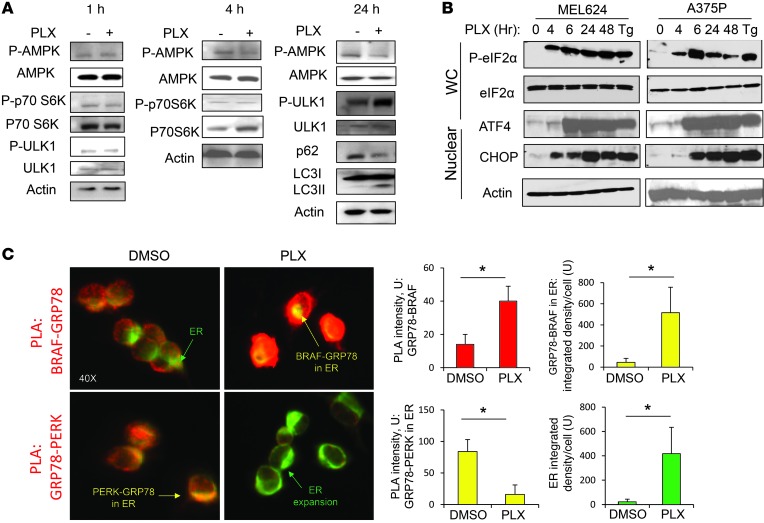
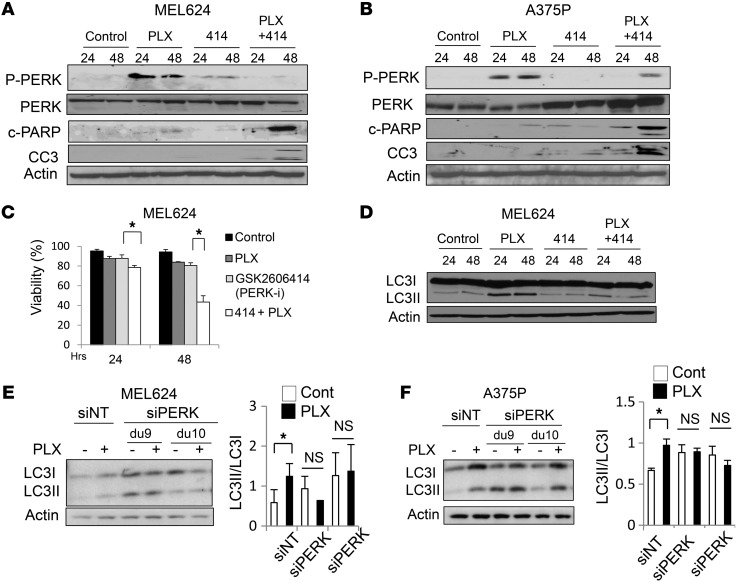
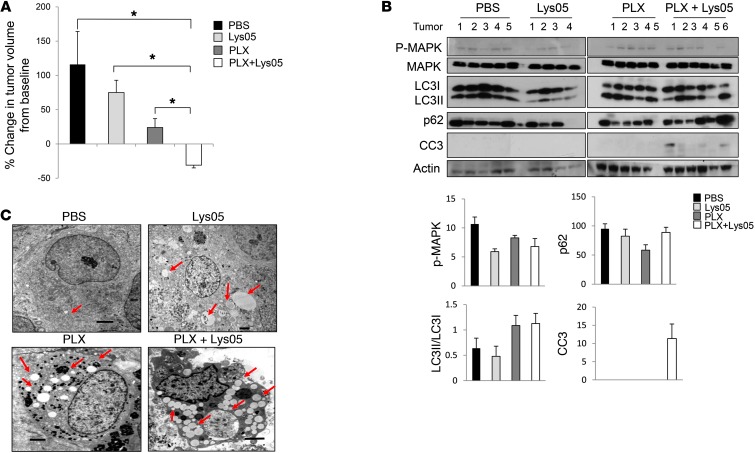
Melanomas that result from mutations in the gene encoding BRAF often become resistant to BRAF inhibition (BRAFi), with multiple mechanisms contributing to resistance. While therapy-induced autophagy promotes resistance to a number of therapies, especially those that target PI3K/mTOR signaling, its role as an adaptive resistance mechanism to BRAFi is not well characterized. Using tumor biopsies from BRAF(V600E) melanoma patients treated either with BRAFi or with combined BRAF and MEK inhibition, we found that BRAFi-resistant tumors had increased levels of autophagy compared with baseline. Patients with higher levels of therapy-induced autophagy had drastically lower response rates to BRAFi and a shorter duration of progression-free survival. In BRAF(V600E) melanoma cell lines, BRAFi or BRAF/MEK inhibition induced cytoprotective autophagy, and autophagy inhibition enhanced BRAFi-induced cell death. Shortly after BRAF inhibitor treatment in melanoma cell lines, mutant BRAF bound the ER stress gatekeeper GRP78, which rapidly expanded the ER. Disassociation of GRP78 from the PKR-like ER-kinase (PERK) promoted a PERK-dependent ER stress response that subsequently activated cytoprotective autophagy. Combined BRAF and autophagy inhibition promoted tumor regression in BRAFi-resistant xenografts. These data identify a molecular pathway for drug resistance connecting BRAFi, the ER stress response, and autophagy and provide a rationale for combination approaches targeting this resistance pathway.
Figures
Comment in
-
Dangerous liaisons: flirtations between oncogenic BRAF and GRP78 in drug-resistant melanomas.J Clin Invest. 2014 Mar;124(3):973-6. doi: 10.1172/JCI74609. Epub 2014 Feb 24. J Clin Invest. 2014. PMID: 24569370 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
