

A frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical Cα-H hydrogen bonds
- PMID: 24569864
- PMCID: PMC3956187
- DOI: 10.1073/pnas.1319944111
A frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical Cα-H hydrogen bonds
Abstract
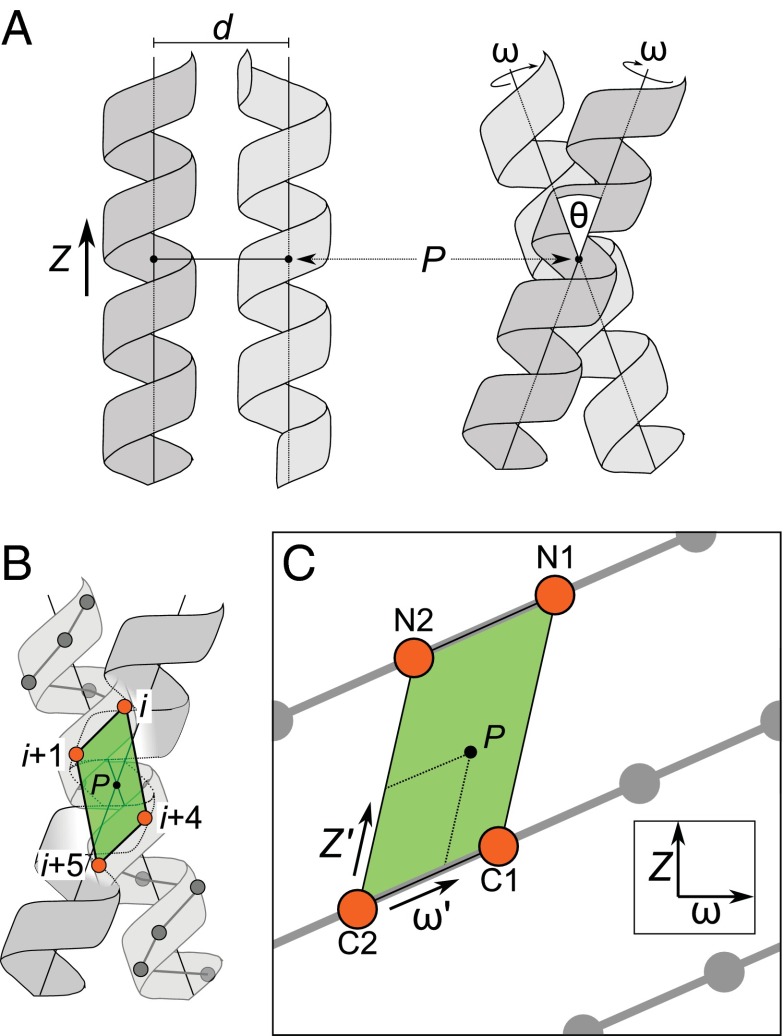
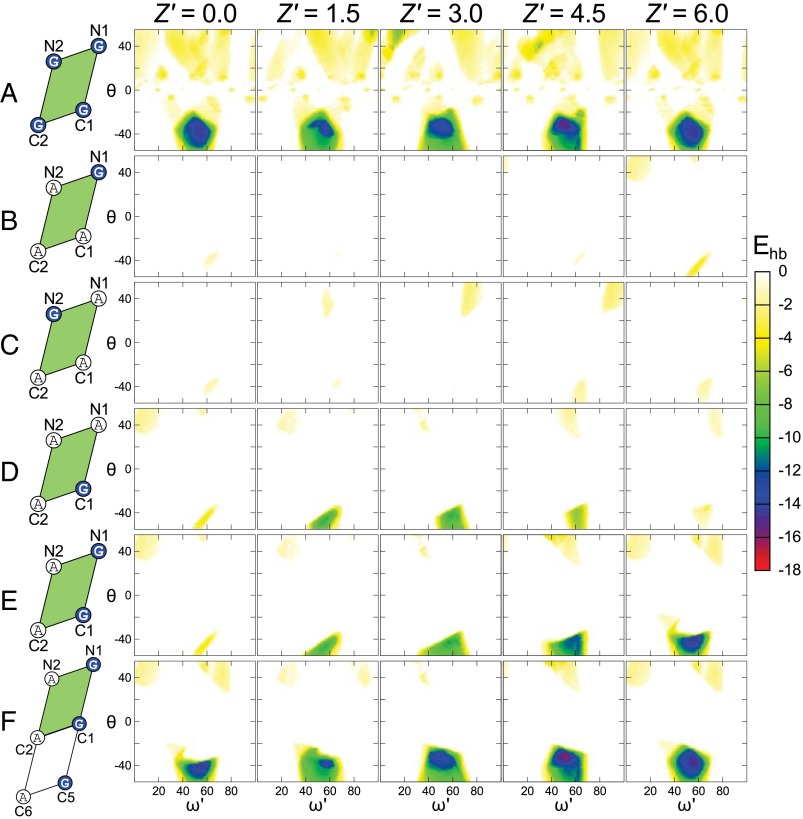
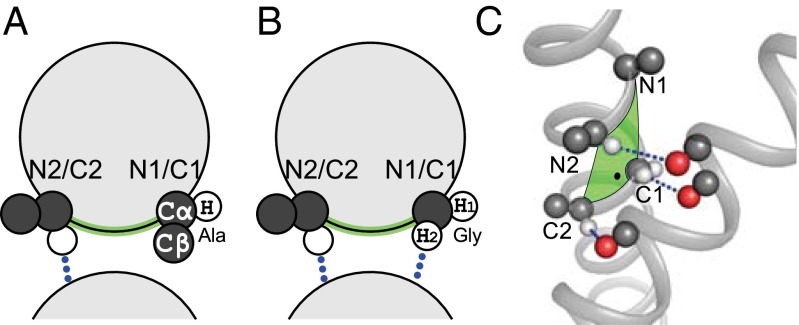
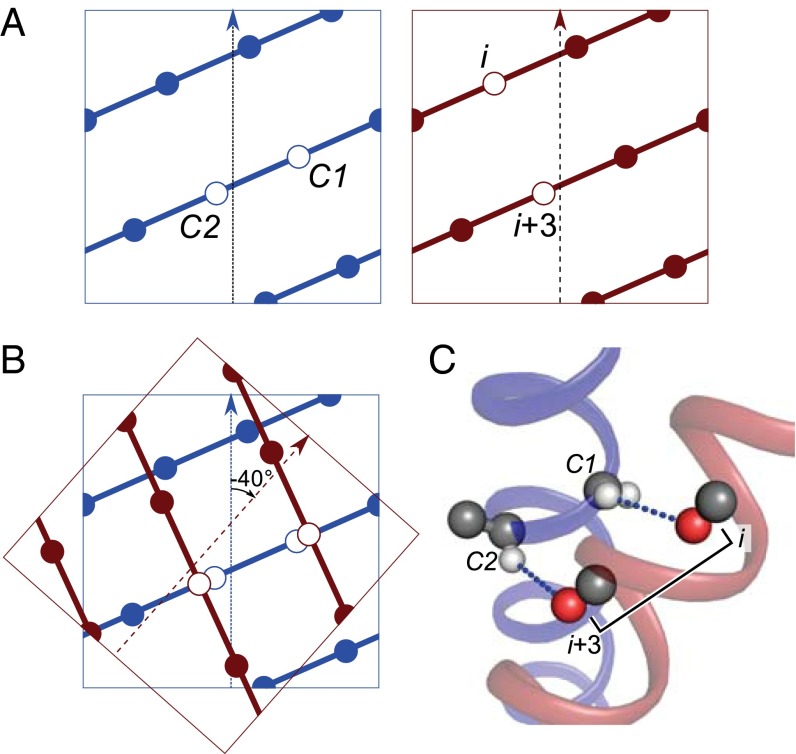
Carbon hydrogen bonds between Cα-H donors and carbonyl acceptors are frequently observed between transmembrane helices (Cα-H···O=C). Networks of these interactions occur often at helix-helix interfaces mediated by GxxxG and similar patterns. Cα-H hydrogen bonds have been hypothesized to be important in membrane protein folding and association, but evidence that they are major determinants of helix association is still lacking. Here we present a comprehensive geometric analysis of homodimeric helices that demonstrates the existence of a single region in conformational space with high propensity for Cα-H···O=C hydrogen bond formation. This region corresponds to the most frequent motif for parallel dimers, GASright, whose best-known example is glycophorin A. The finding suggests a causal link between the high frequency of occurrence of GASright and its propensity for carbon hydrogen bond formation. Investigation of the sequence dependency of the motif determined that Gly residues are required at specific positions where only Gly can act as a donor with its "side chain" Hα. Gly also reduces the steric barrier for non-Gly amino acids at other positions to act as Cα donors, promoting the formation of cooperative hydrogen bonding networks. These findings offer a structural rationale for the occurrence of GxxxG patterns at the GASright interface. The analysis identified the conformational space and the sequence requirement of Cα-H···O=C mediated motifs; we took advantage of these results to develop a structural prediction method. The resulting program, CATM, predicts ab initio the known high-resolution structures of homodimeric GASright motifs at near-atomic level.
Keywords: interaction motifs; protein prediction.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
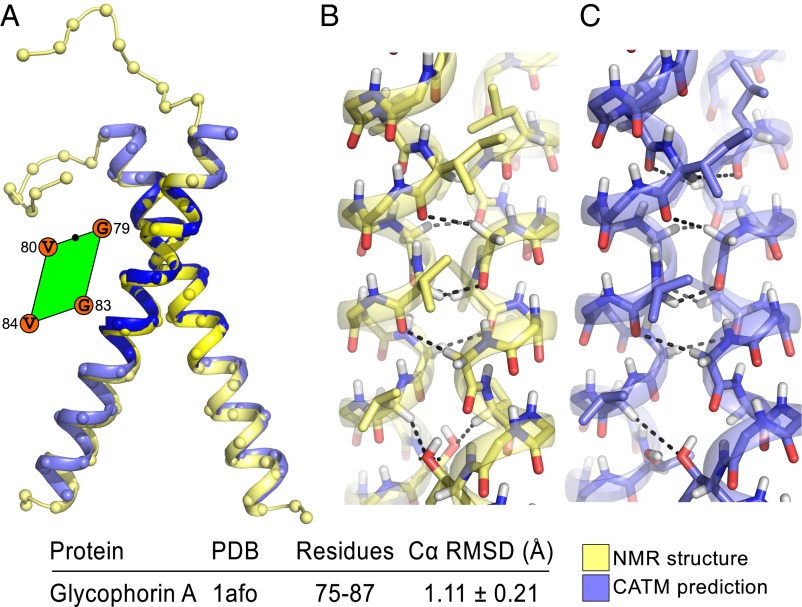
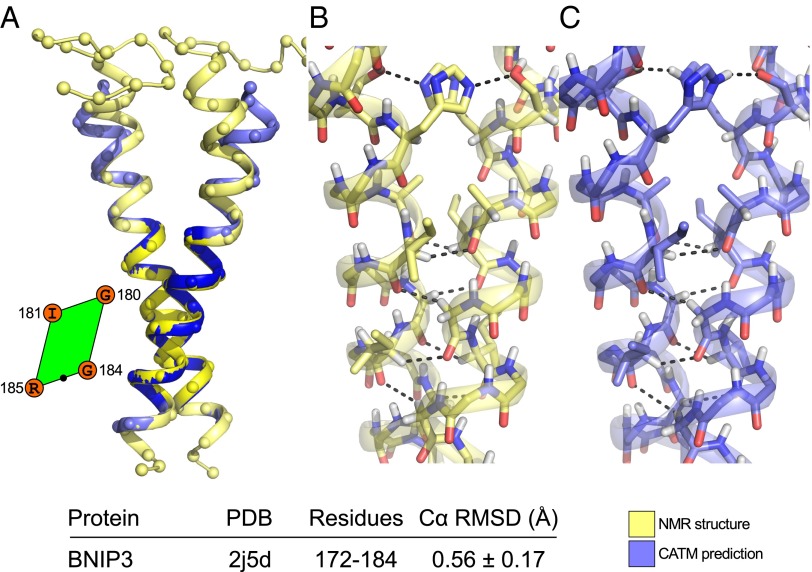
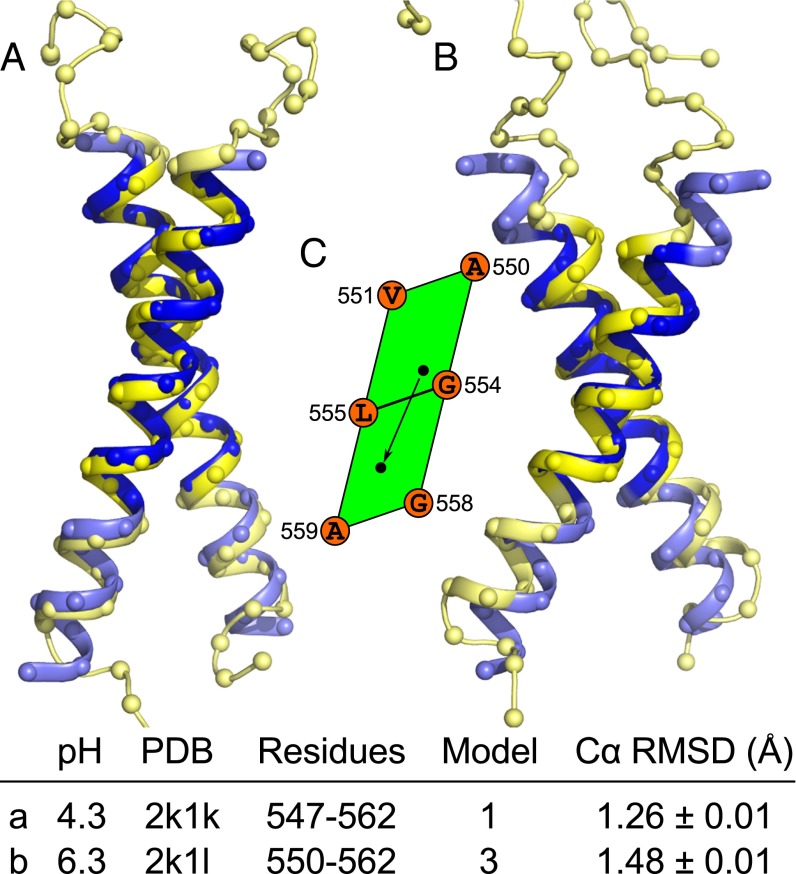
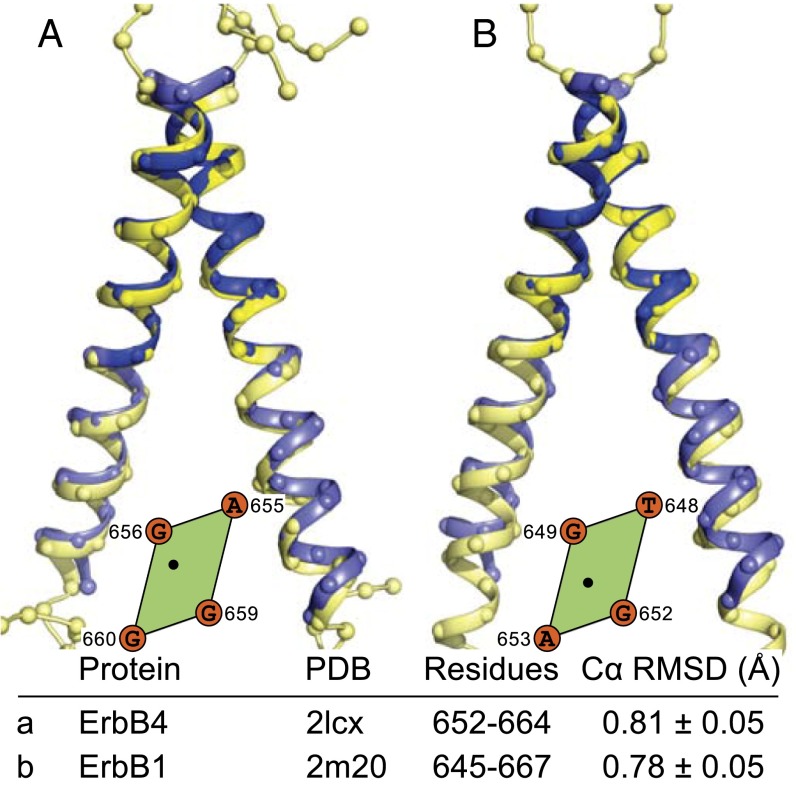
References
-
- Senes A, Gerstein M, Engelman DM. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J Mol Biol. 2000;296(3):921–936. - PubMed
-
- Lomize MA, Lomize AL, Pogozheva ID, Mosberg HI. OPM: Orientations of Proteins in Membranes database. Bioinformatics. 2006;22(5):623–625. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
