

Bifunctional homodimeric triokinase/FMN cyclase: contribution of protein domains to the activities of the human enzyme and molecular dynamics simulation of domain movements
- PMID: 24569995
- PMCID: PMC4036181
- DOI: 10.1074/jbc.M113.525626
Bifunctional homodimeric triokinase/FMN cyclase: contribution of protein domains to the activities of the human enzyme and molecular dynamics simulation of domain movements
Abstract
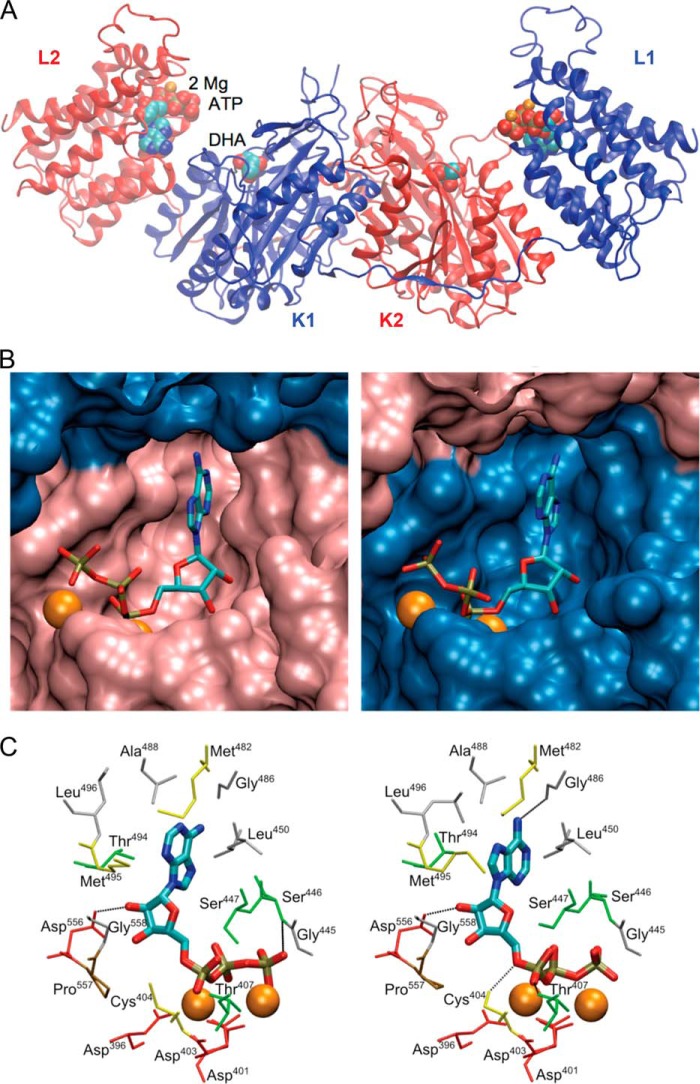
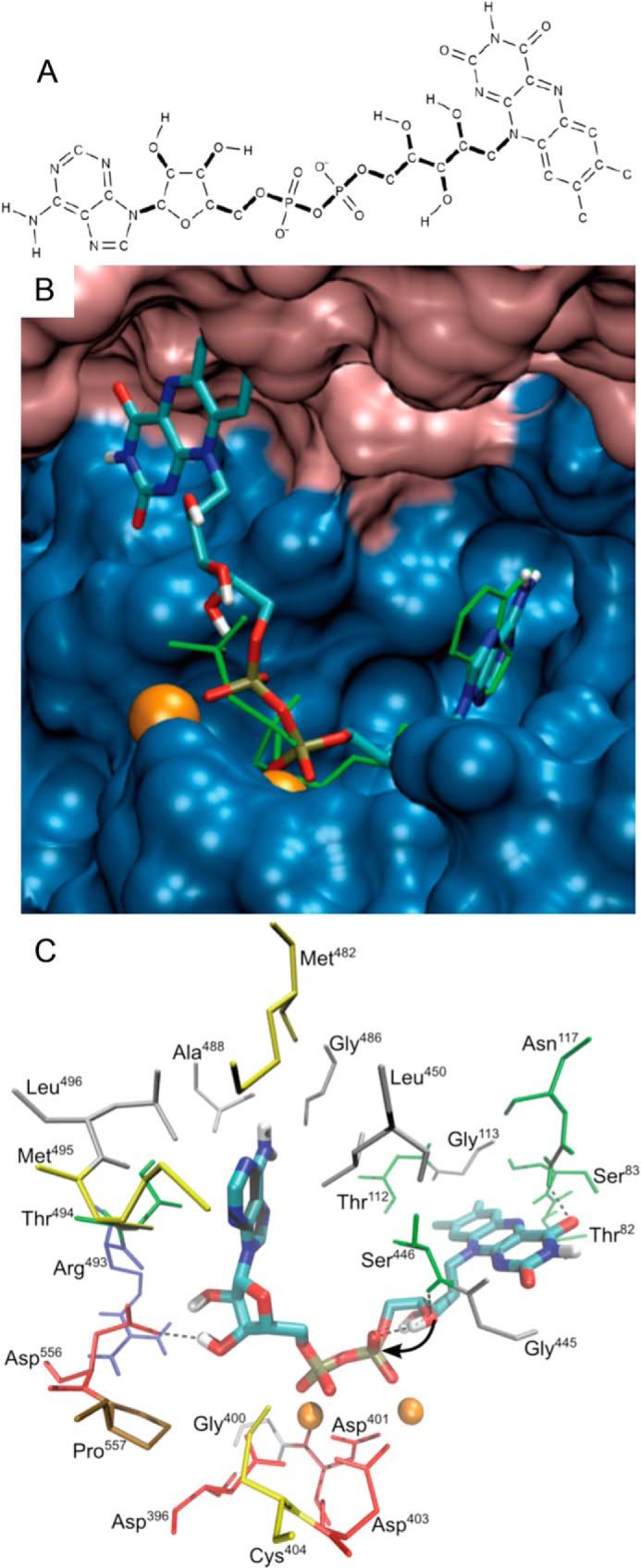
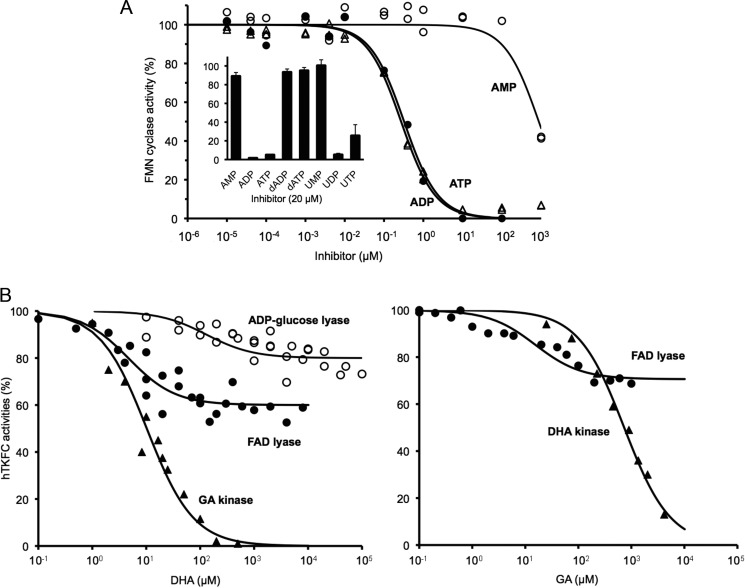
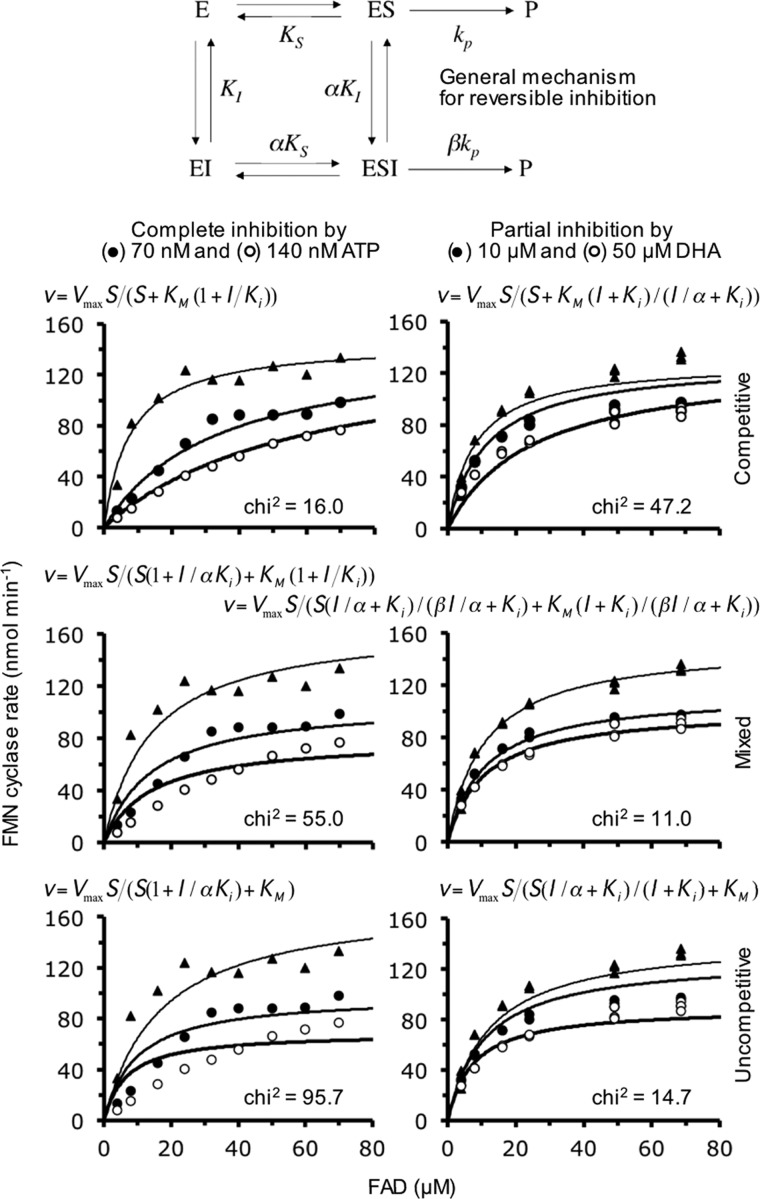
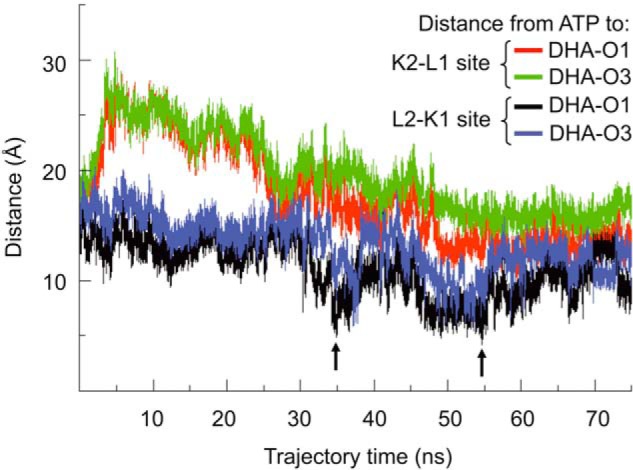
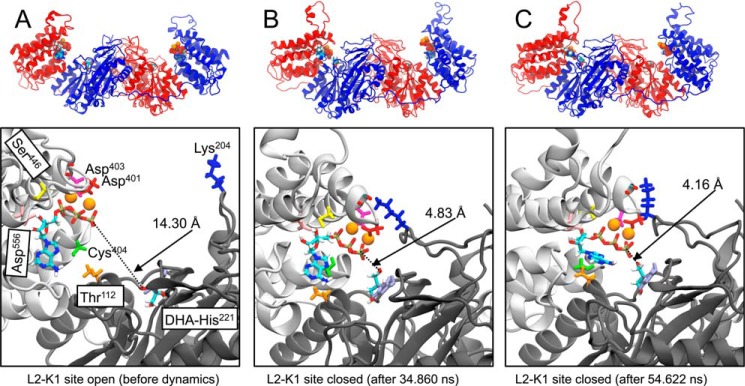
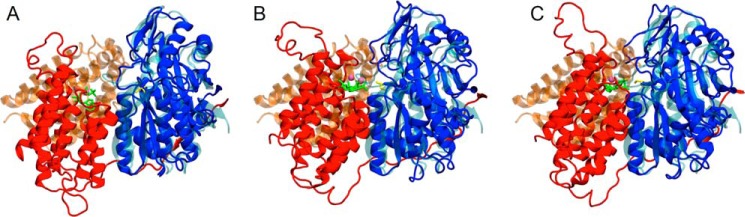
Mammalian triokinase, which phosphorylates exogenous dihydroxyacetone and fructose-derived glyceraldehyde, is neither molecularly identified nor firmly associated to an encoding gene. Human FMN cyclase, which splits FAD and other ribonucleoside diphosphate-X compounds to ribonucleoside monophosphate and cyclic X-phosphodiester, is identical to a DAK-encoded dihydroxyacetone kinase. This bifunctional protein was identified as triokinase. It was modeled as a homodimer of two-domain (K and L) subunits. Active centers lie between K1 and L2 or K2 and L1: dihydroxyacetone binds K and ATP binds L in different subunits too distant (≈ 14 Å) for phosphoryl transfer. FAD docked to the ATP site with ribityl 4'-OH in a possible near-attack conformation for cyclase activity. Reciprocal inhibition between kinase and cyclase reactants confirmed substrate site locations. The differential roles of protein domains were supported by their individual expression: K was inactive, and L displayed cyclase but not kinase activity. The importance of domain mobility for the kinase activity of dimeric triokinase was highlighted by molecular dynamics simulations: ATP approached dihydroxyacetone at distances below 5 Å in near-attack conformation. Based upon structure, docking, and molecular dynamics simulations, relevant residues were mutated to alanine, and kcat and Km were assayed whenever kinase and/or cyclase activity was conserved. The results supported the roles of Thr(112) (hydrogen bonding of ATP adenine to K in the closed active center), His(221) (covalent anchoring of dihydroxyacetone to K), Asp(401) and Asp(403) (metal coordination to L), and Asp(556) (hydrogen bonding of ATP or FAD ribose to L domain). Interestingly, the His(221) point mutant acted specifically as a cyclase without kinase activity.
Keywords: Bifunctional Kinase/Cyclase; Cyclic FMN; DAK Gene; Dihydroxyacetone Phosphorylation; FAD; Fructose Metabolism; Glyceraldehyde Phosphorylation; Molecular Docking; Molecular Dynamics; Mutagenesis, Site-specific.
Figures
References
-
- Hers H. G., Kusaka T. (1953) The metabolism of fructose-1-phosphate in the liver. Biochim. Biophys. Acta 11, 427–437 - PubMed
-
- Sillero M. A., Sillero A., Sols A. (1969) Enzymes involved in fructose metabolism in liver and the glyceraldehyde metabolic crossroads. Eur. J. Biochem. 10, 345–350 - PubMed
-
- Mayes P. A. (1993) Intermediary metabolism of fructose. Am. J. Clin. Nutr. 58, 754S–765S - PubMed
-
- Steinmann B., Gitzelmann R., Van den Berghe G. (2001) in The Metabolic and Molecular Basis of Inherited Disease (Scriver C., Beaudet A., Sly W., Valle D., Childs B., Vogelstein B., eds) 8th Ed., pp. 1489–1520, McGraw-Hill, New York
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
