

Prospects for neuroprotective therapies in prodromal Huntington's disease
- PMID: 24573776
- PMCID: PMC4624289
- DOI: 10.1002/mds.25835
Prospects for neuroprotective therapies in prodromal Huntington's disease
Abstract
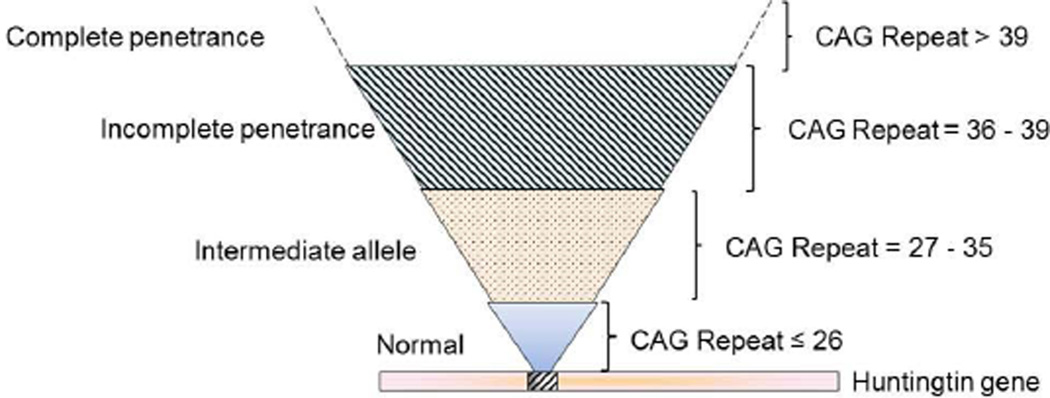
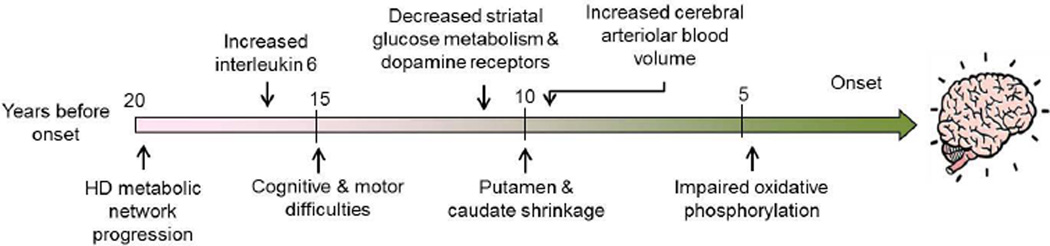
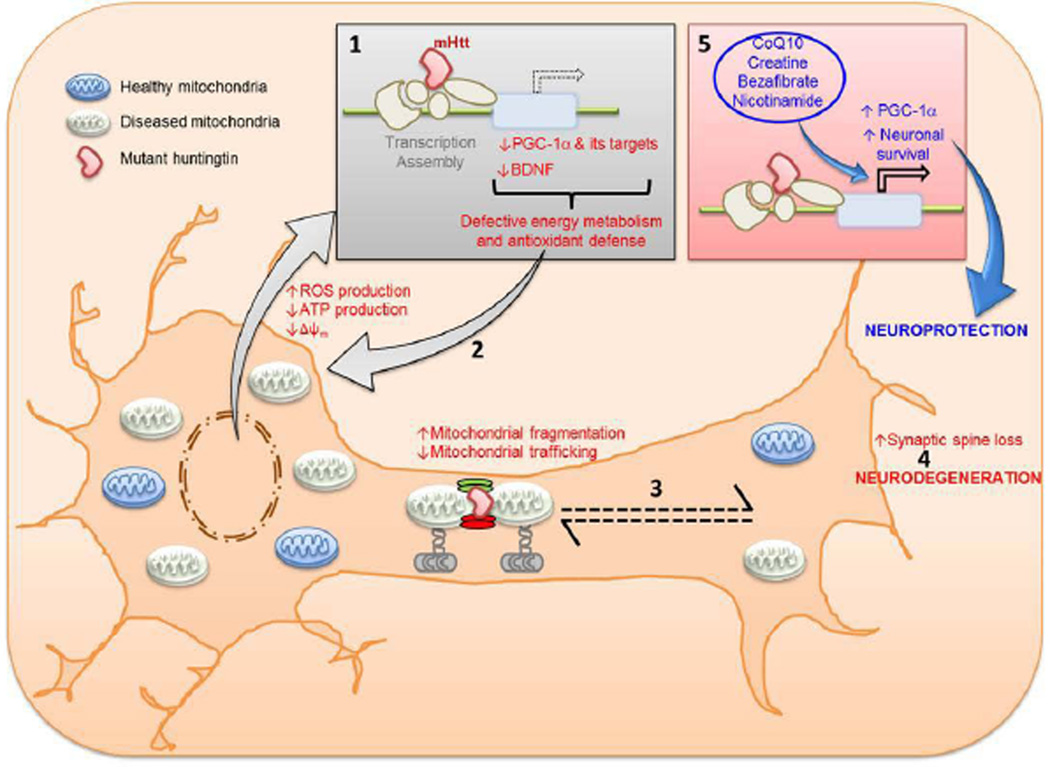
Huntington's disease (HD) is a prototypical dominantly inherited neurodegenerative disorder characterized by progressive cognitive deterioration, psychiatric disturbances, and a movement disorder. The genetic cause of the illness is a CAG repeat expansion in the huntingtin gene, which leads to a polyglutamine expansion in the huntingtin protein. The exact mechanism by which mutant huntingtin causes HD is unknown, but it causes abnormalities in gene transcription as well as both mitochondrial dysfunction and oxidative damage. Because the penetrance of HD is complete with CAG repeats greater than 39, patients can be diagnosed well before disease onset with genetic testing. Longitudinal studies of HD patients before disease onset have shown that subtle cognitive and motor deficits occur as much as 10 years before onset, as do reductions in glucose utilization and striatal atrophy. An increase in inflammation, as shown by elevated interleukin-6, occurs approximately 15 years before onset. Detection of these abnormalities may be useful in defining an optimal time for disease intervention to try to slow or halt the degenerative process. Although reducing gene expression with small interfering RNA or short hairpin RNA is an attractive approach, other approaches targeting energy metabolism, inflammation, and oxidative damage may be more easily and rapidly moved into the clinic. The recent PREQUEL study of coenzyme Q10 in presymptomatic gene carriers showed the feasibility of carrying out clinical trials to slow or halt onset of HD. We review both the earliest detectable clinical and laboratory manifestations of HD, as well as potential neuroprotective therapies that could be utilized in presymptomatic HD.
Keywords: PGC-1alpha; SIRT1; bezafibrate; coenzyme Q10; presymptomatic Huntington's disease.
© 2014 International Parkinson and Movement Disorder Society.
Conflict of interest statement
Authors declare no conflict of interest
Figures
Similar articles
-
The role of mitochondrial dysfunction in Huntington's disease: Implications for therapeutic targeting.Biomed Pharmacother. 2025 Feb;183:117827. doi: 10.1016/j.biopha.2025.117827. Epub 2025 Jan 23. Biomed Pharmacother. 2025. PMID: 39854819 Review.
-
Mitochondrial and metabolic-based protective strategies in Huntington's disease: the case of creatine and coenzyme Q.Rev Neurosci. 2011 Dec 2;23(1):13-28. doi: 10.1515/RNS.2011.060. Rev Neurosci. 2011. PMID: 22150069 Review.
-
Neuroprotective effect of solanesol against 3-nitropropionic acid-induced Huntington's disease-like behavioral, biochemical, and cellular alterations: Restoration of coenzyme-Q10-mediated mitochondrial dysfunction.Indian J Pharmacol. 2018 Nov-Dec;50(6):309-319. doi: 10.4103/ijp.IJP_11_18. Indian J Pharmacol. 2018. PMID: 30783323 Free PMC article.
-
Drp1/Fis1-mediated mitochondrial fragmentation leads to lysosomal dysfunction in cardiac models of Huntington's disease.J Mol Cell Cardiol. 2019 Feb;127:125-133. doi: 10.1016/j.yjmcc.2018.12.004. Epub 2018 Dec 11. J Mol Cell Cardiol. 2019. PMID: 30550751 Free PMC article.
-
Antioxidants in Huntington's disease.Biochim Biophys Acta. 2012 May;1822(5):664-74. doi: 10.1016/j.bbadis.2011.11.014. Epub 2011 Nov 23. Biochim Biophys Acta. 2012. PMID: 22138129 Free PMC article. Review.
Cited by
-
Preclinical progression of neurodegenerative diseases.Nagoya J Med Sci. 2018 Aug;80(3):289-298. doi: 10.18999/nagjms.80.3.289. Nagoya J Med Sci. 2018. PMID: 30214078 Free PMC article. Review.
-
Impaired muscle uptake of creatine in spinal and bulbar muscular atrophy.Ann Clin Transl Neurol. 2016 Jun 23;3(7):537-46. doi: 10.1002/acn3.324. eCollection 2016 Jul. Ann Clin Transl Neurol. 2016. PMID: 27386502 Free PMC article.
-
Utility of the Huntington's Disease Prognostic Index Score for a Perimanifest Clinical Trial.Mov Disord. 2022 May;37(5):1040-1046. doi: 10.1002/mds.28944. Epub 2022 Feb 16. Mov Disord. 2022. PMID: 35170086 Free PMC article.
-
Planning for Prevention of Parkinson Disease: Now Is the Time.Neurology. 2022 Aug 16;99(7 Suppl 1):1-9. doi: 10.1212/WNL.0000000000200789. Neurology. 2022. PMID: 36219787 Free PMC article.
-
A common gene expression signature in Huntington's disease patient brain regions.BMC Med Genomics. 2014 Oct 30;7:60. doi: 10.1186/s12920-014-0060-2. BMC Med Genomics. 2014. PMID: 25358814 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
