

Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling
- PMID: 24574393
- PMCID: PMC3993837
- DOI: 10.1128/JVI.03851-13
Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling
Abstract
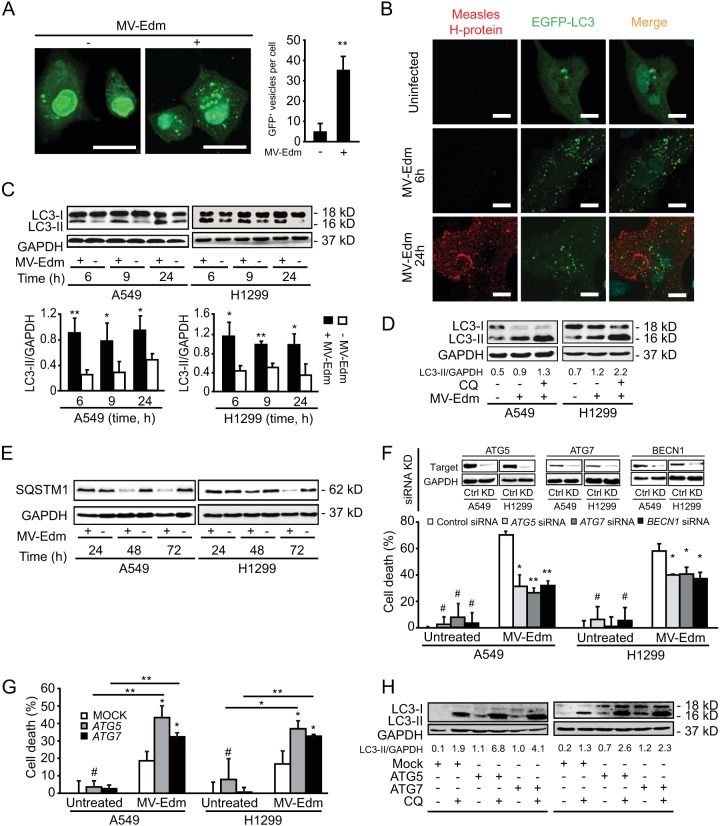
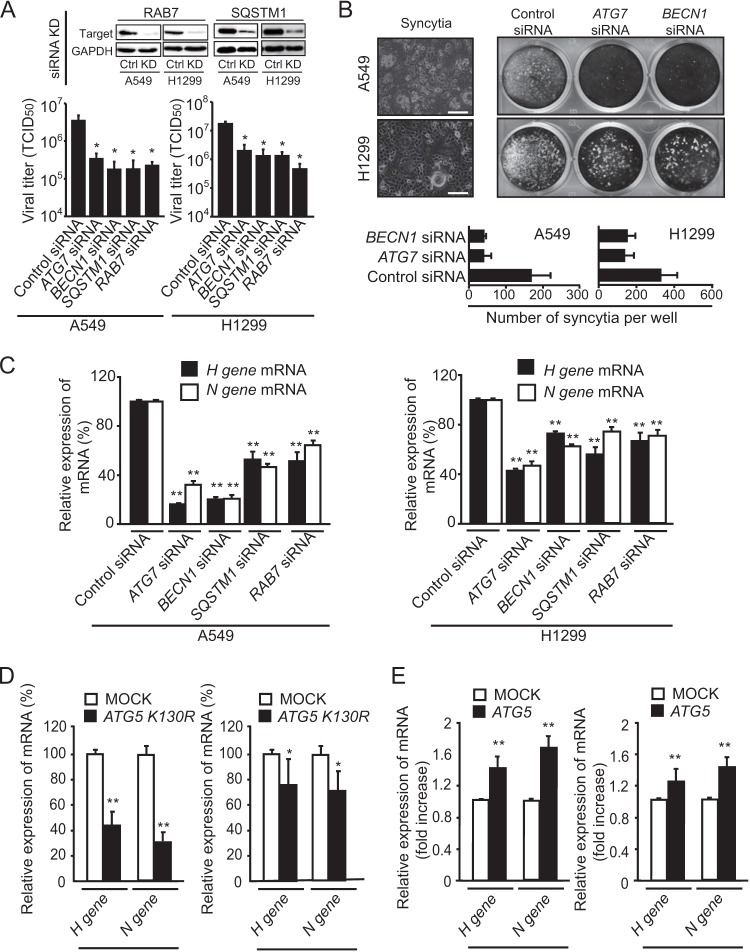
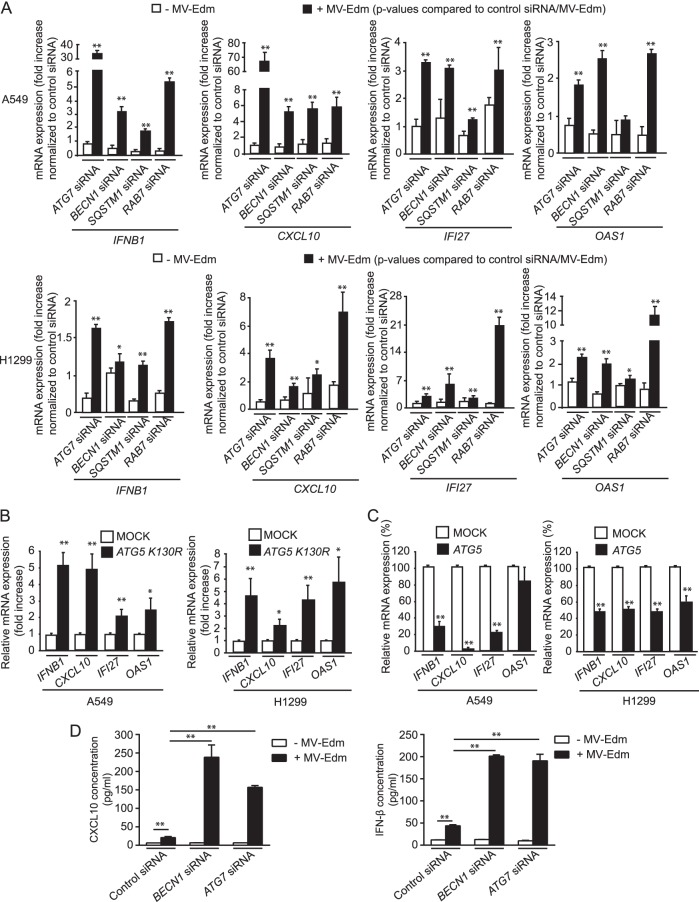
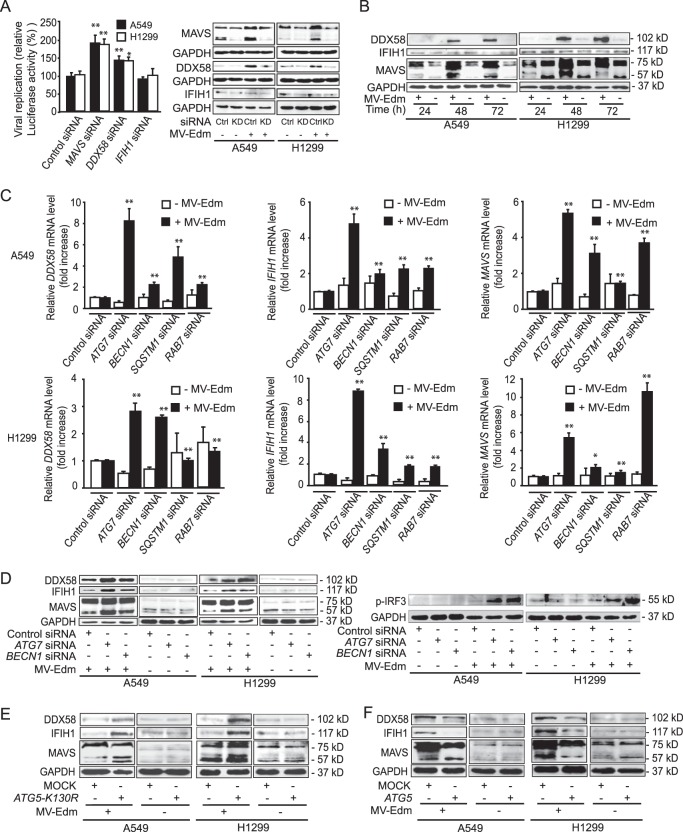
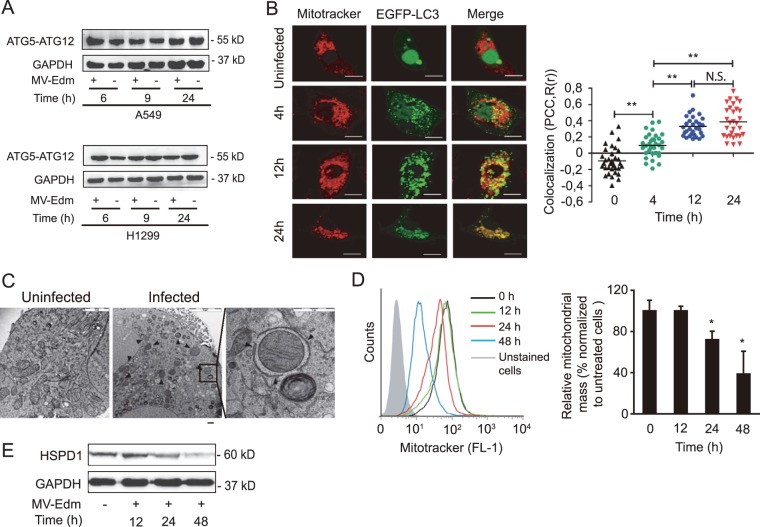
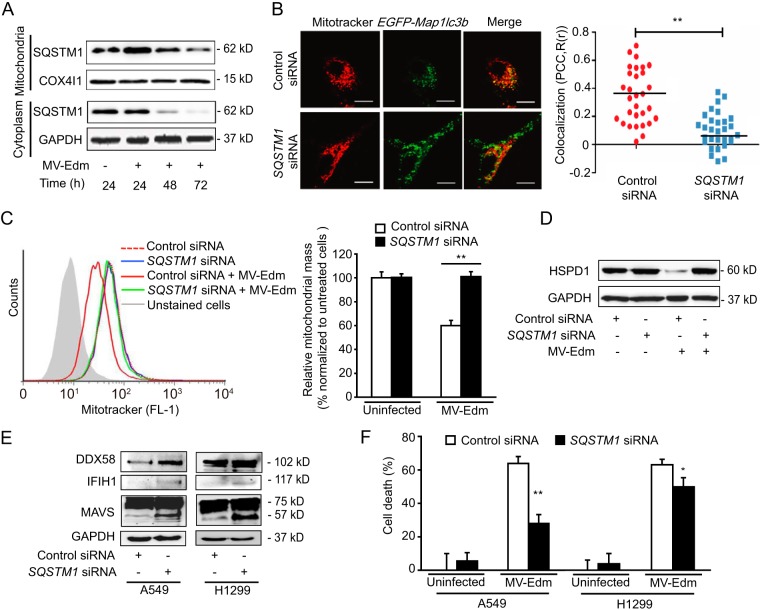
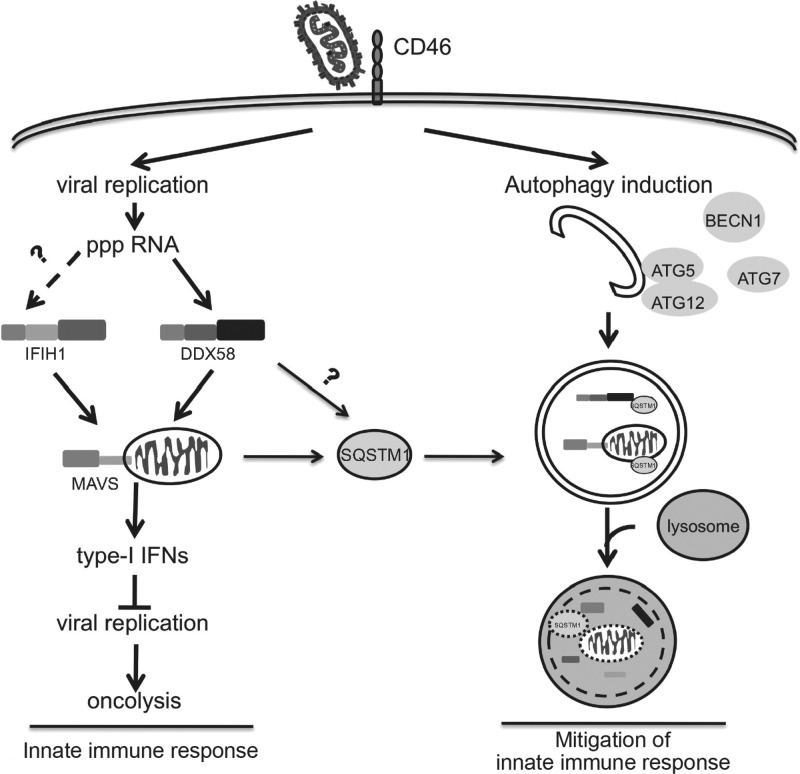
The success of future clinical trials with oncolytic viruses depends on the identification and the control of mechanisms that modulate their therapeutic efficacy. In particular, little is known about the role of autophagy in infection by attenuated measles virus of the Edmonston strain (MV-Edm). We investigated the interaction between autophagy, innate immune response, and oncolytic activity of MV-Edm, since the antiviral immune response is a known factor limiting virotherapies. We report that MV-Edm exploits selective autophagy to mitigate the innate immune response mediated by DDX58/RIG-I like receptors (RLRs) in non-small cell lung cancer (NSCLC) cells. Both RNA interference (RNAi) and overexpression approaches demonstrate that autophagy enhances viral replication and inhibits the production of type I interferons regulated by RLRs. We show that MV-Edm unexpectedly triggers SQSTM1/p62-mediated mitophagy, resulting in decreased mitochondrion-tethered mitochondrial antiviral signaling protein (MAVS) and subsequently weakening the innate immune response. These results unveil a novel infectious strategy based on the usurpation of mitophagy leading to mitigation of the innate immune response. This finding provides a rationale to modulate autophagy in oncolytic virotherapy.
Importance: In vitro studies, preclinical experiments in vivo, and clinical trials with humans all indicate that oncolytic viruses hold promise for cancer therapy. Measles virus of the Edmonston strain (MV-Edm), which is an attenuated virus derived from the common wild-type measles virus, is paradigmatic for therapeutic oncolytic viruses. MV-Edm replicates preferentially in and kills cancer cells. The efficiency of MV-Edm is limited by the immune response of the host against viruses. In our study, we revealed that MV-Edm usurps a homeostatic mechanism of intracellular degradation of mitochondria, coined mitophagy, to attenuate the innate immune response in cancer cells. This strategy might provide a replicative advantage for the virus against the development of antiviral immune responses by the host. These findings are important since they may not only indicate that inducers of autophagy could enhance the efficacy of oncolytic therapies but also provide clues for antiviral therapy by targeting SQSTM1/p62-mediated mitophagy.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
