

Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer
- PMID: 24574398
- PMCID: PMC4019131
- DOI: 10.1128/JVI.03688-13
Complementary induction of immunogenic cell death by oncolytic parvovirus H-1PV and gemcitabine in pancreatic cancer
Abstract
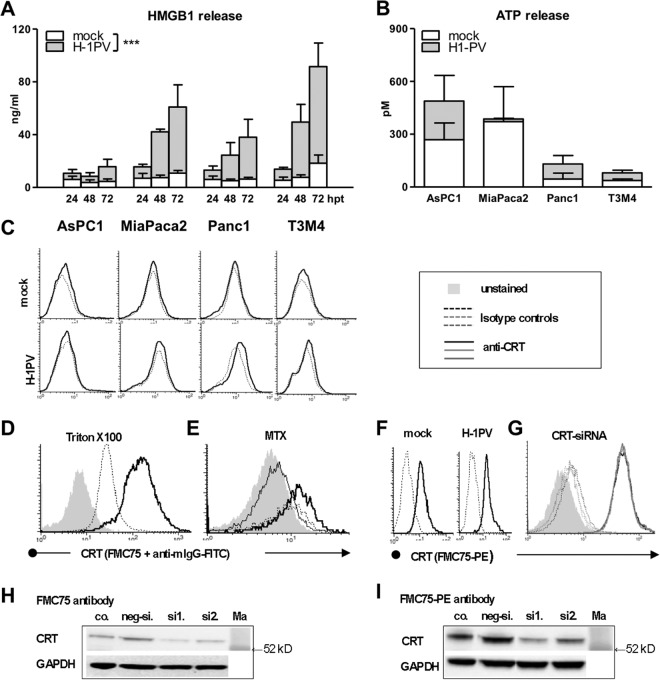
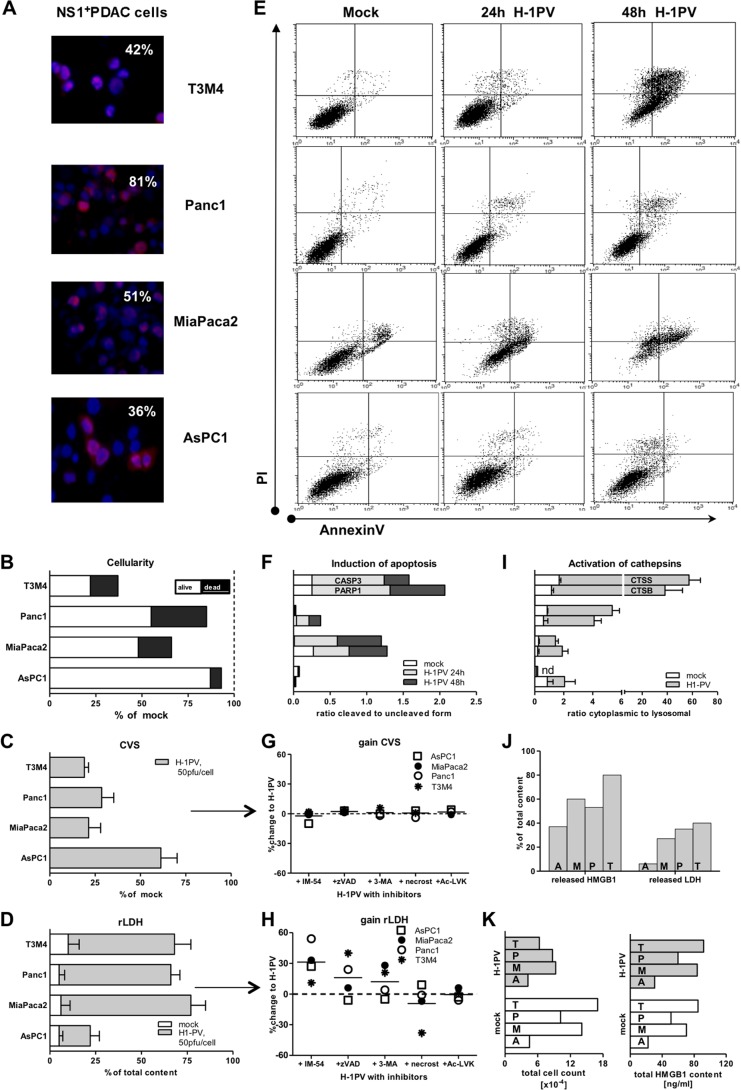
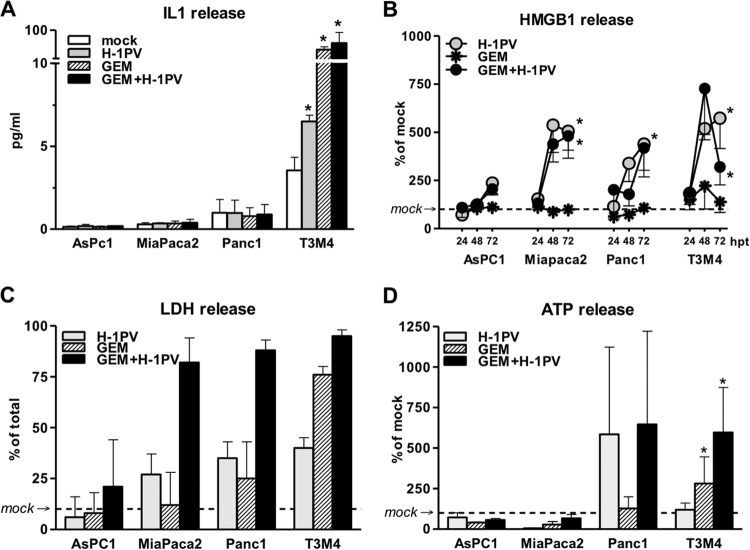
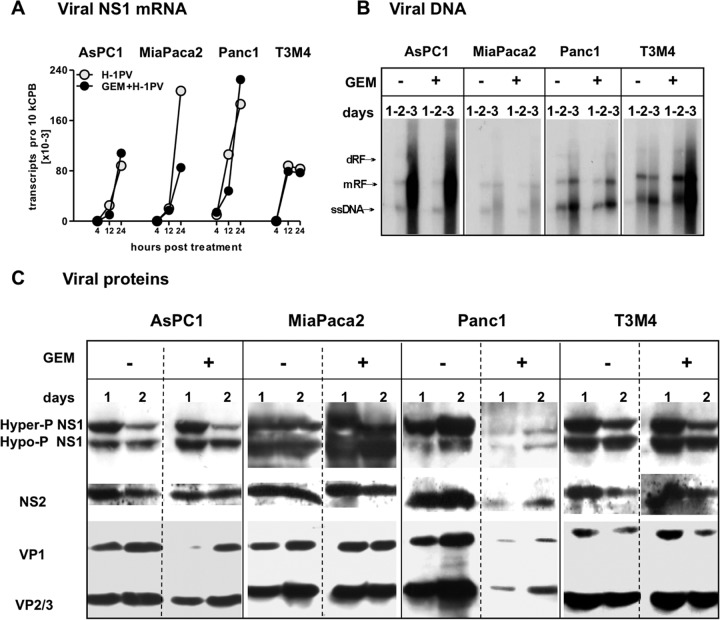
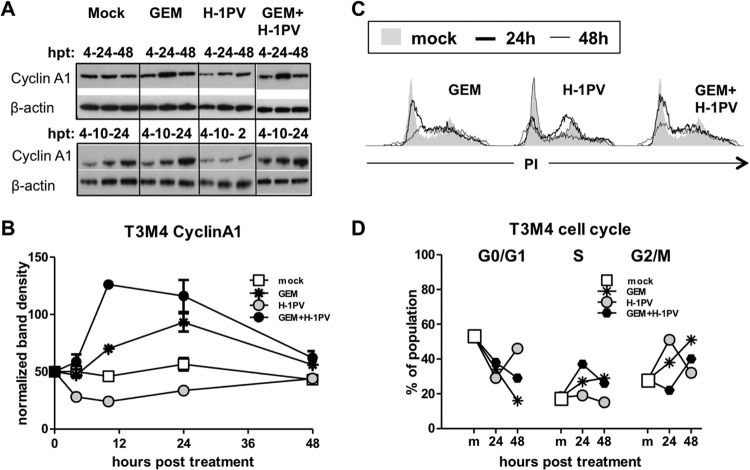
Novel therapies employing oncolytic viruses have emerged as promising anticancer modalities. The cure of particularly aggressive malignancies requires induction of immunogenic cell death (ICD), coupling oncolysis with immune responses via calreticulin, ATP, and high-mobility group box protein B1 (HMGB1) release from dying tumor cells. The present study shows that in human pancreatic cancer cells (pancreatic ductal adenocarcinoma [PDAC] cells n=4), oncolytic parvovirus H-1 (H-1PV) activated multiple interconnected death pathways but failed to induce calreticulin exposure or ATP release. In contrast, H-1PV elevated extracellular HMGB1 levels by 4.0±0.5 times (58%±9% of total content; up to 100 ng/ml) in all infected cultures, whether nondying, necrotic, or apoptotic. An alternative secretory route allowed H-1PV to overcome the failure of gemcitabine to trigger HMGB1 release, without impeding cytotoxicity or other ICD activities of the standard PDAC medication. Such broad resistance of H-1PV-induced HMGB1 release to apoptotic blockage coincided with but was uncoupled from an autocrine interleukin-1β (IL-1β) loop. That and the pattern of viral determinants maintained in gemcitabine-treated cells suggested the activation of an inflammasome/caspase 1 (CASP1) platform alongside DNA detachment and/or nuclear exclusion of HMGB1 during early stages of the viral life cycle. We concluded that H-1PV infection of PDAC cells is signaled through secretion of the alarmin HMGB1 and, besides its own oncolytic effect, might convert drug-induced apoptosis into an ICD process. A transient arrest of cells in the cyclin A1-rich S phase would suffice to support compatibility of proliferation-dependent H-1PV with cytotoxic regimens. These properties warrant incorporation of the oncolytic virus H-1PV, which is not pathogenic in humans, into multimodal anticancer treatments.
Importance: The current therapeutic concepts targeting aggressive malignancies require an induction of immunogenic cell death characterized by exposure of calreticulin (CRT) as well as release of ATP and HMGB1 from dying cells. In pancreatic tumor cells (PDAC cells) infected with the oncolytic parvovirus H-1PV, only HMGB1 was released by all infected cells, whether nondying, necrotic, or succumbing to one of the programmed death pathways, including contraproductive apoptosis. Our data suggest that active secretion of HMGB1 from PDAC cells is a sentinel reaction emerging during early stages of the viral life cycle, irrespective of cell death, that is compatible with and complements cytotoxic regimens. Consistent induction of HMGB1 secretion raised the possibility that this reaction might be a general "alarming" phenomenon characteristic of H-1PV's interaction with the host cell; release of IL-1β points to the possible involvement of a danger-sensing inflammasome platform. Both provide a basis for further virus-oriented studies.
Figures
References
-
- Igney FH, Krammer PH. 2002. Immune escape of tumors: apoptosis resistance and tumor counterattack. J. Leukoc. Biol. 71:907–920 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
