

Akt and mTOR mediate programmed necrosis in neurons
- PMID: 24577082
- PMCID: PMC3944276
- DOI: 10.1038/cddis.2014.69
Akt and mTOR mediate programmed necrosis in neurons
Abstract
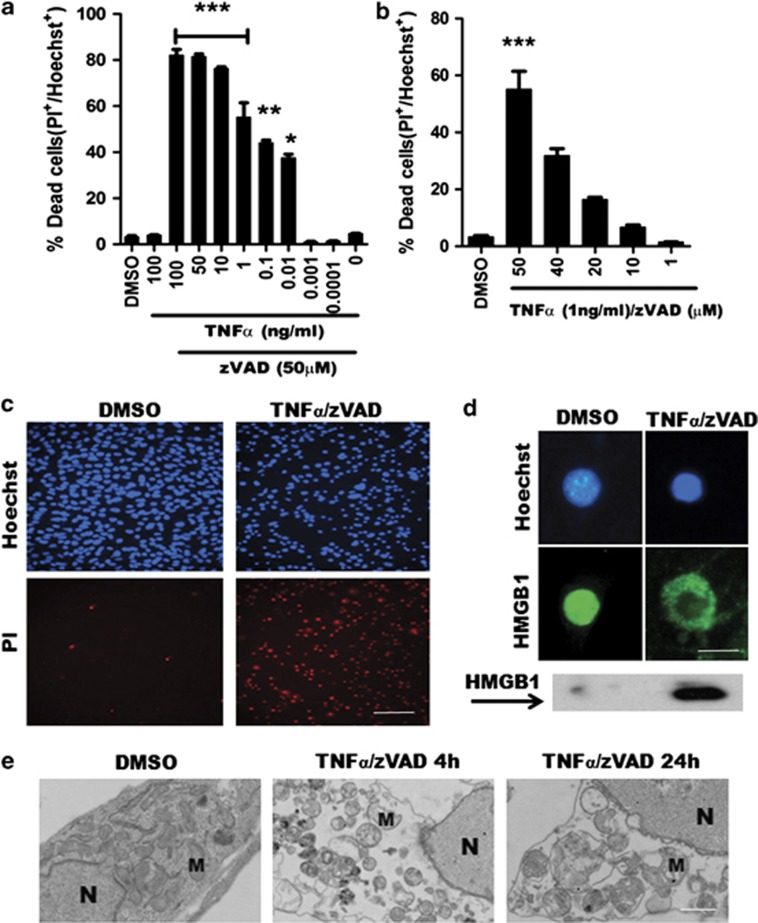
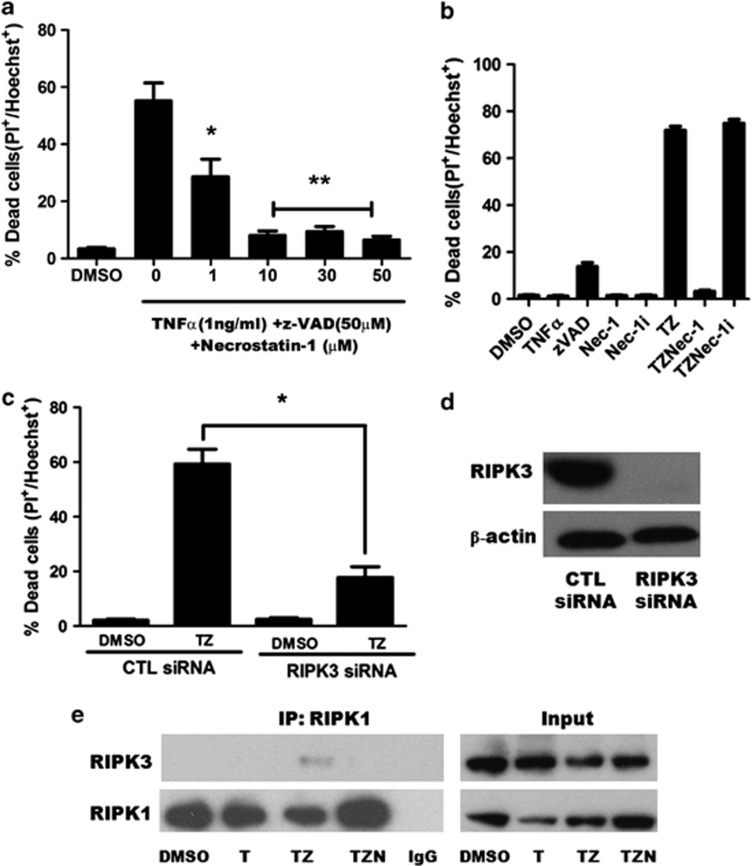
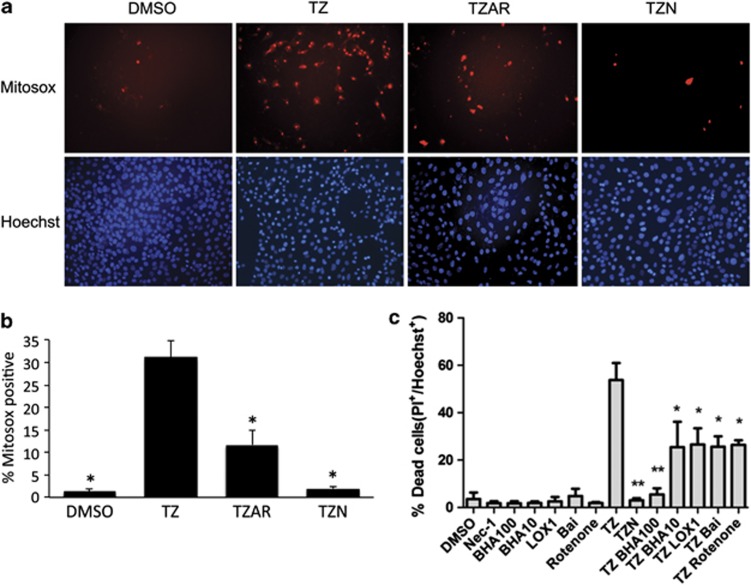
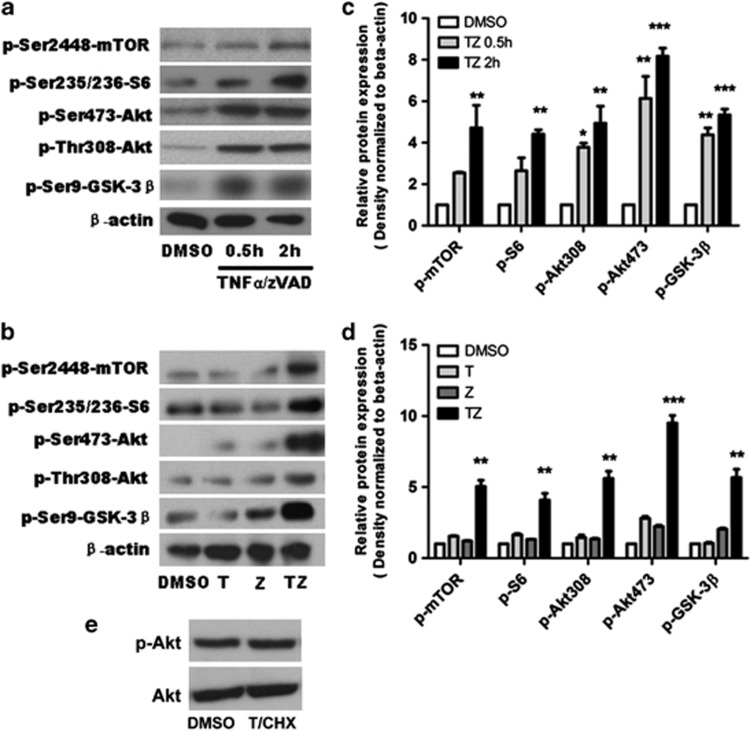
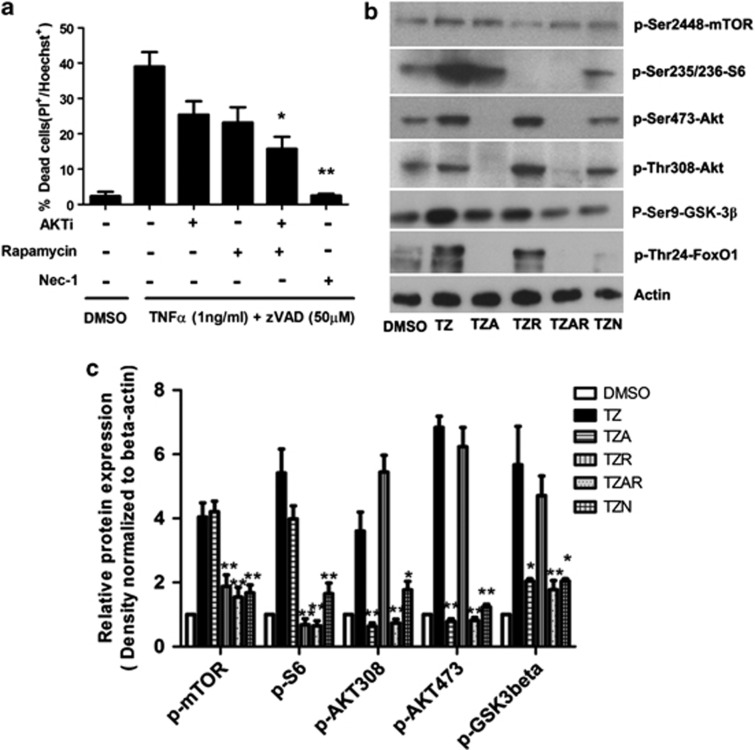
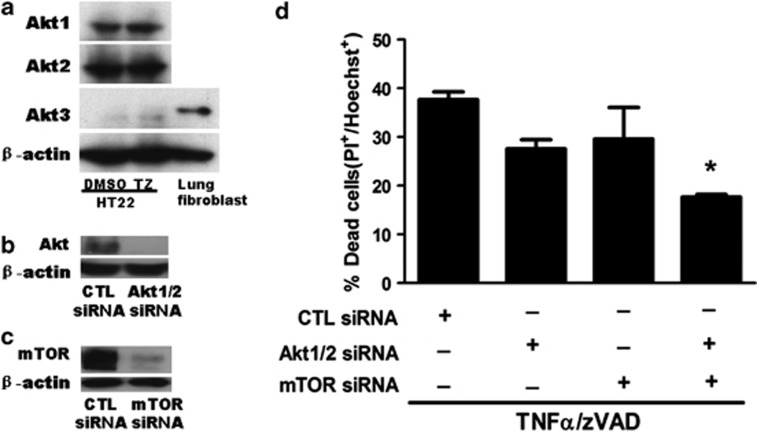
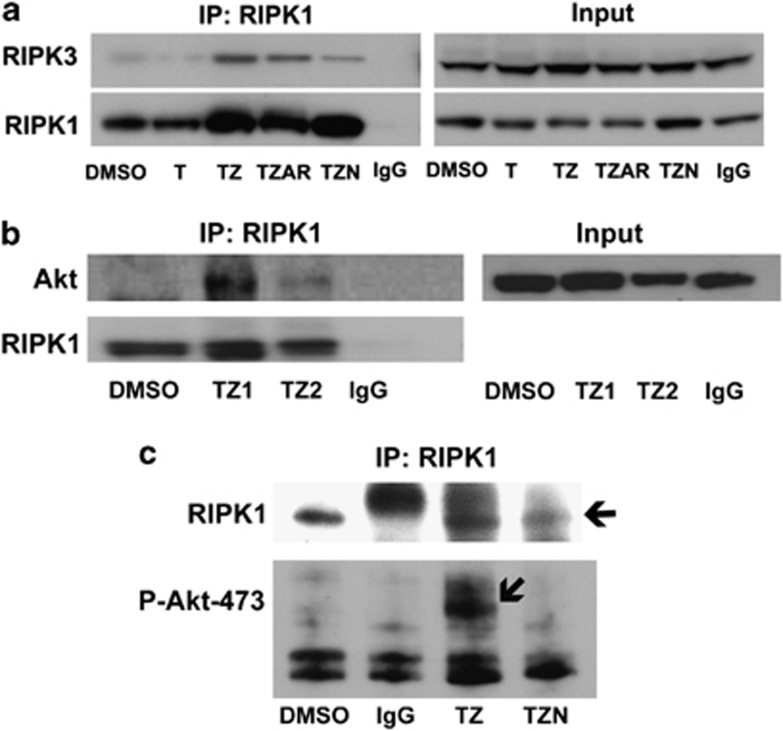
Necroptosis is a newly described form of regulated necrosis that contributes to neuronal death in experimental models of stroke and brain trauma. Although much work has been done elucidating initiating mechanisms, signaling events governing necroptosis remain largely unexplored. Akt is known to inhibit apoptotic neuronal cell death. Mechanistic target of rapamycin (mTOR) is a downstream effector of Akt that controls protein synthesis. We previously reported that dual inhibition of Akt and mTOR reduced acute cell death and improved long term cognitive deficits after controlled-cortical impact in mice. These findings raised the possibility that Akt/mTOR might regulate necroptosis. To test this hypothesis, we induced necroptosis in the hippocampal neuronal cell line HT22 using concomitant treatment with tumor necrosis factor α (TNFα) and the pan-caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone. TNFα/zVAD treatment induced cell death within 4 h. Cell death was preceded by RIPK1-RIPK3-pAkt assembly, and phosphorylation of Thr-308 and Thr473 of AKT and its direct substrate glycogen synthase kinase-3β, as well as mTOR and its direct substrate S6 ribosomal protein (S6), suggesting activation of Akt/mTOR pathways. Pretreatment with Akt inhibitor viii and rapamycin inhibited Akt and S6 phosphorylation events, mitochondrial reactive oxygen species production, and necroptosis by over 50% without affecting RIPK1-RIPK3 complex assembly. These data were confirmed using small inhibitory ribonucleic acid-mediated knockdown of AKT1/2 and mTOR. All of the aforementioned biochemical events were inhibited by necrostatin-1, including Akt and mTOR phosphorylation, generation of oxidative stress, and RIPK1-RIPK3-pAkt complex assembly. The data suggest a novel, heretofore unexpected role for Akt and mTOR downstream of RIPK1 activation in neuronal cell death.
Figures
Similar articles
-
Critical contribution of oxidative stress to TNFα-induced necroptosis downstream of RIPK1 activation.Biochem Biophys Res Commun. 2013 Jun 28;436(2):212-6. doi: 10.1016/j.bbrc.2013.05.075. Epub 2013 May 29. Biochem Biophys Res Commun. 2013. PMID: 23727581
-
RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis.FEBS J. 2014 Nov;281(21):4921-34. doi: 10.1111/febs.13034. Epub 2014 Oct 4. FEBS J. 2014. PMID: 25195660
-
Aβ-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis.Biochim Biophys Acta. 2016 Nov;1863(11):2820-2834. doi: 10.1016/j.bbamcr.2016.09.003. Epub 2016 Sep 4. Biochim Biophys Acta. 2016. PMID: 27599716
-
The regulation of necroptosis and perspectives for the development of new drugs preventing ischemic/reperfusion of cardiac injury.Apoptosis. 2022 Oct;27(9-10):697-719. doi: 10.1007/s10495-022-01760-x. Epub 2022 Aug 20. Apoptosis. 2022. PMID: 35986803 Review.
-
Taking aim at Alzheimer's disease through the mammalian target of rapamycin.Ann Med. 2014 Dec;46(8):587-96. doi: 10.3109/07853890.2014.941921. Epub 2014 Aug 8. Ann Med. 2014. PMID: 25105207 Free PMC article. Review.
Cited by
-
Targeted PI3K/AKT-hyperactivation induces cell death in chronic lymphocytic leukemia.Nat Commun. 2021 Jun 10;12(1):3526. doi: 10.1038/s41467-021-23752-2. Nat Commun. 2021. PMID: 34112805 Free PMC article.
-
mTOR in Alzheimer disease and its earlier stages: Links to oxidative damage in the progression of this dementing disorder.Free Radic Biol Med. 2021 Jun;169:382-396. doi: 10.1016/j.freeradbiomed.2021.04.025. Epub 2021 Apr 30. Free Radic Biol Med. 2021. PMID: 33933601 Free PMC article. Review.
-
Phenotypic screening identifies a new oxazolone inhibitor of necroptosis and neuroinflammation.Cell Death Discov. 2018 Jul 10;4:10. doi: 10.1038/s41420-018-0067-0. eCollection 2018. Cell Death Discov. 2018. PMID: 30062059 Free PMC article.
-
Targeting TNFα-mediated cytotoxicity using thalidomide after experimental cardiac arrest in rats: An exploratory study.Exp Ther Med. 2022 Jun;23(6):380. doi: 10.3892/etm.2022.11307. Epub 2022 Apr 8. Exp Ther Med. 2022. PMID: 35495588 Free PMC article.
-
A receptor-interacting protein 1 (RIP1)-independent necrotic death under the control of protein phosphatase PP2A that involves the reorganization of actin cytoskeleton and the action of cofilin-1.J Biol Chem. 2014 Sep 12;289(37):25699-710. doi: 10.1074/jbc.M114.575134. Epub 2014 Aug 5. J Biol Chem. 2014. PMID: 25096578 Free PMC article.
References
-
- Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. - PubMed
-
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. - PubMed
-
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
