

ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies
- PMID: 24584572
- PMCID: PMC4240914
- DOI: 10.1007/s00467-013-2706-2
ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies
Abstract
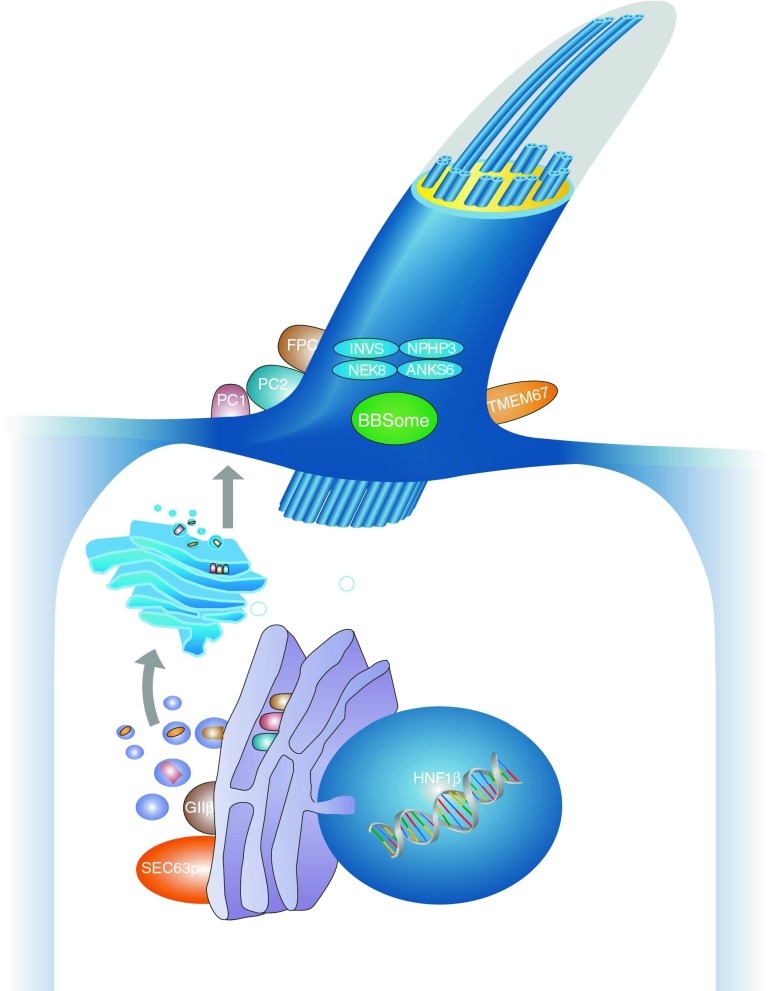
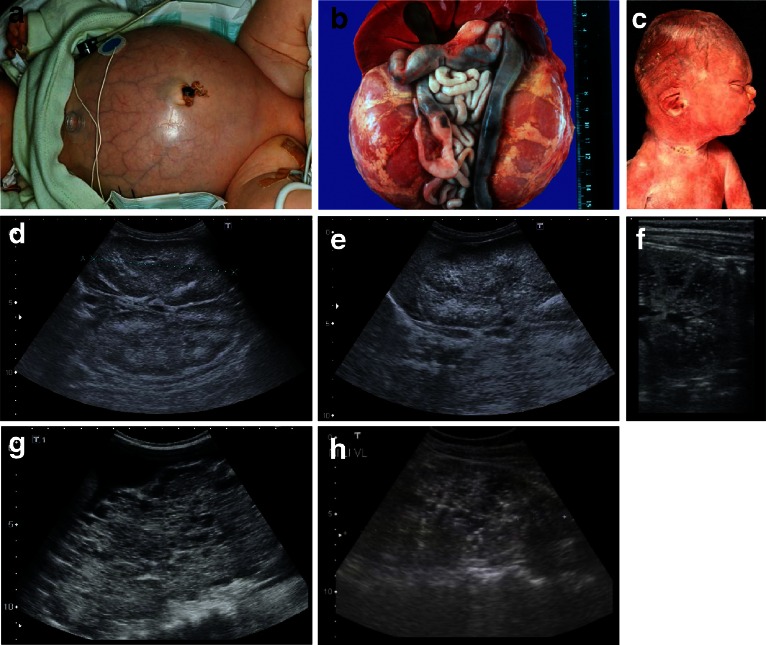
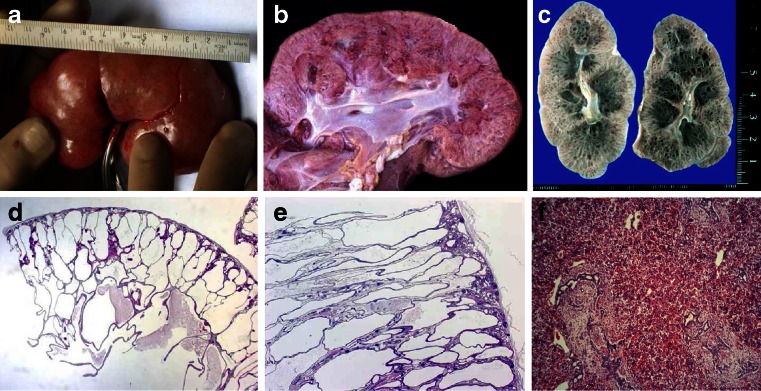
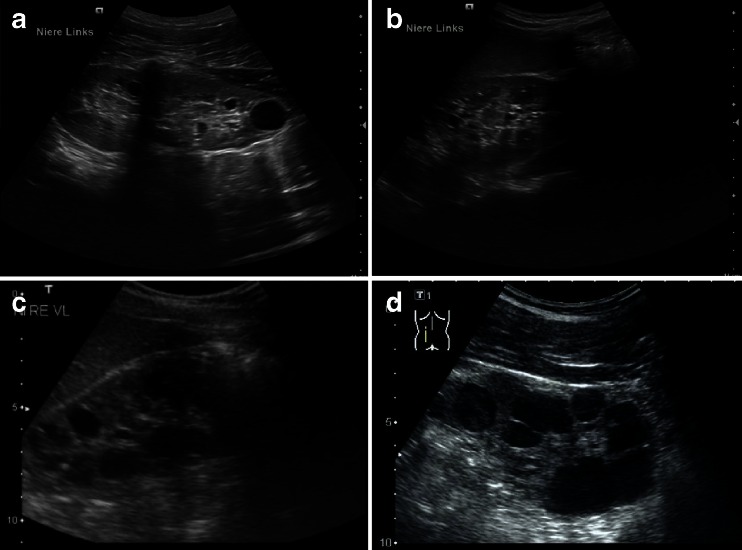
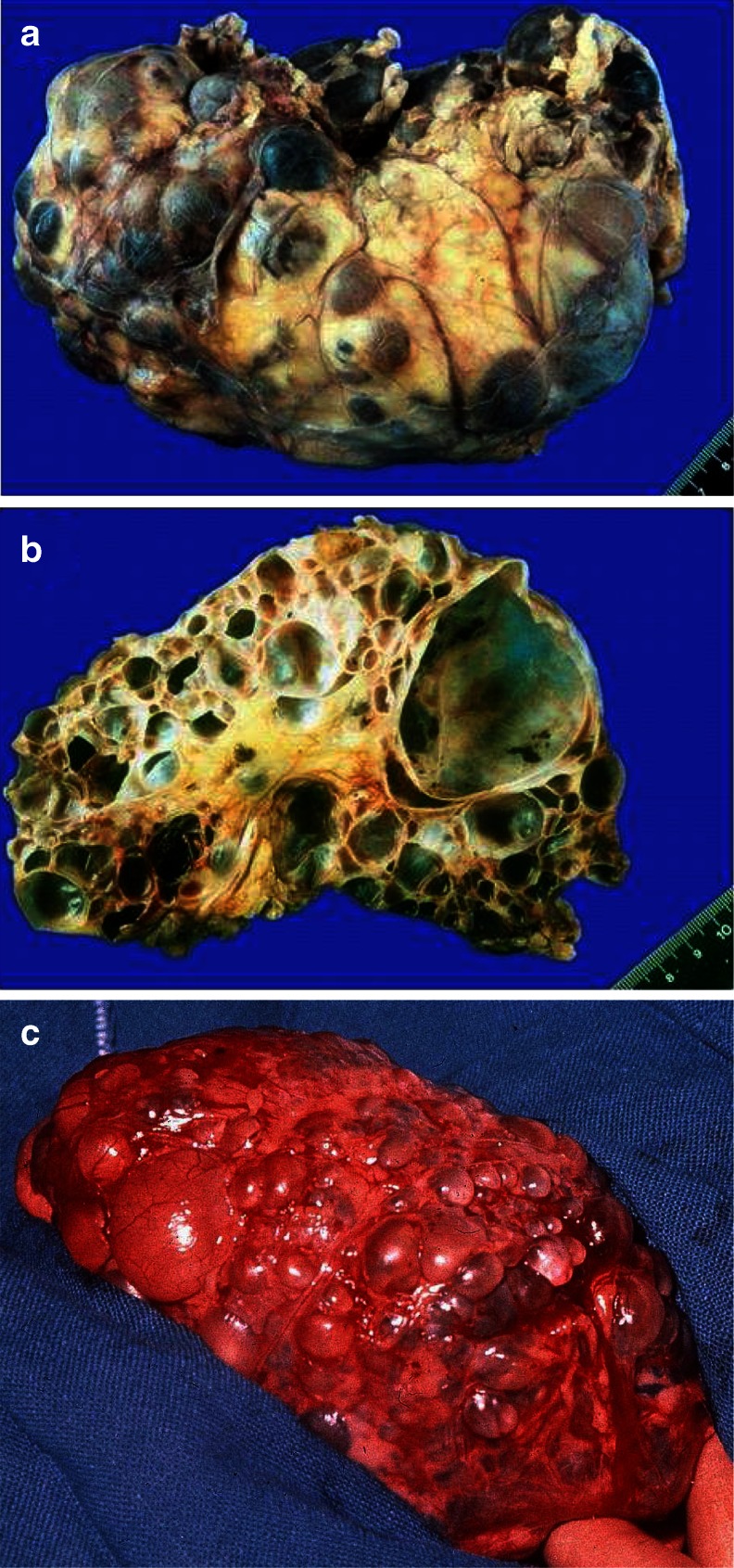
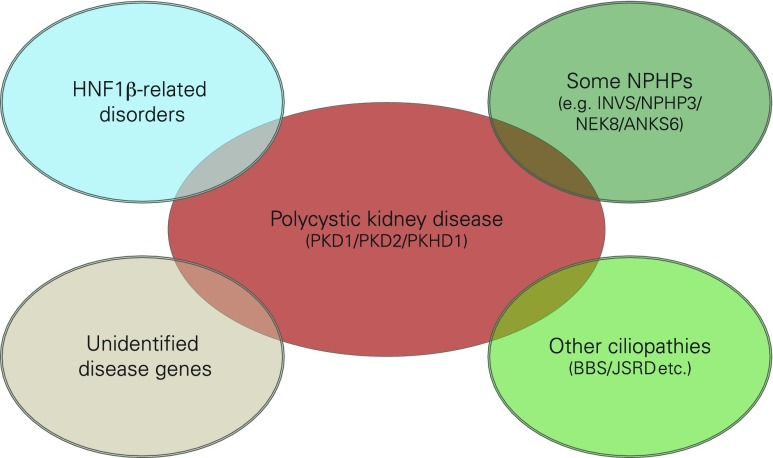
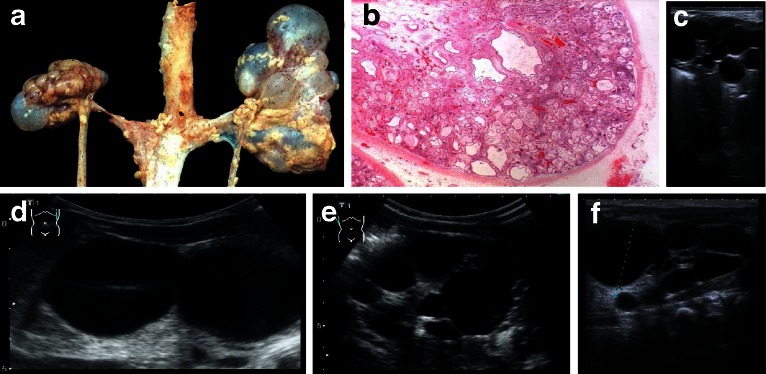
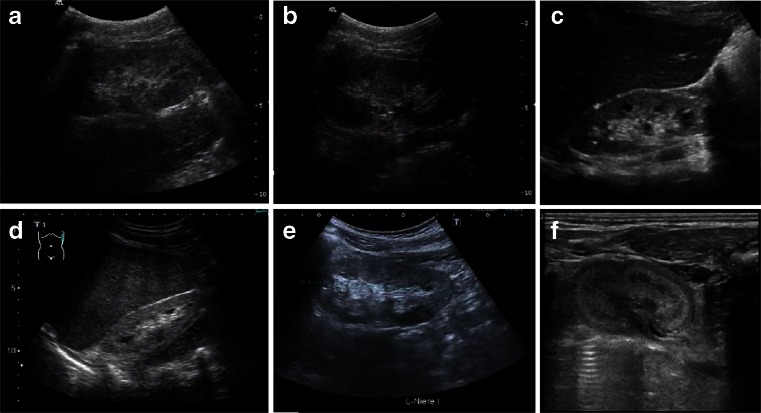
Renal cysts are clinically and genetically heterogeneous conditions. Polycystic kidney disease (PKD) is common and its characterization has paved the way for the identification of a growing number of cilia-related disorders (ciliopathies) of which most show cystic kidneys. While the recessive form of PKD (ARPKD) virtually always presents in childhood, early onset can, in some instances, also occur in the dominant form (ADPKD). Both ADPKD genes (PKD1 and PKD2) can also be inherited in a recessive way, making the story more complex with evidence for a dosage-sensitive network. Several phenocopies are known, and mutations in HNF1ß or genes that typically cause other ciliopathies, such as nephronophthisis, Bardet-Biedl, Joubert syndrome and related disorders, can mimic PKD. An accurate genetic diagnosis is crucial for genetic counseling, prenatal diagnostics, and the clinical management of patients and their families. The increasing number of genes that have to be considered in patients with cystic kidney disease is challenging to address by conventional techniques and largely benefits from next-generation sequencing-based approaches. The parallel analysis of targeted genes considerably increases the detection rate, allows for better interpretation of identified variants, and avoids genetic misdiagnoses.
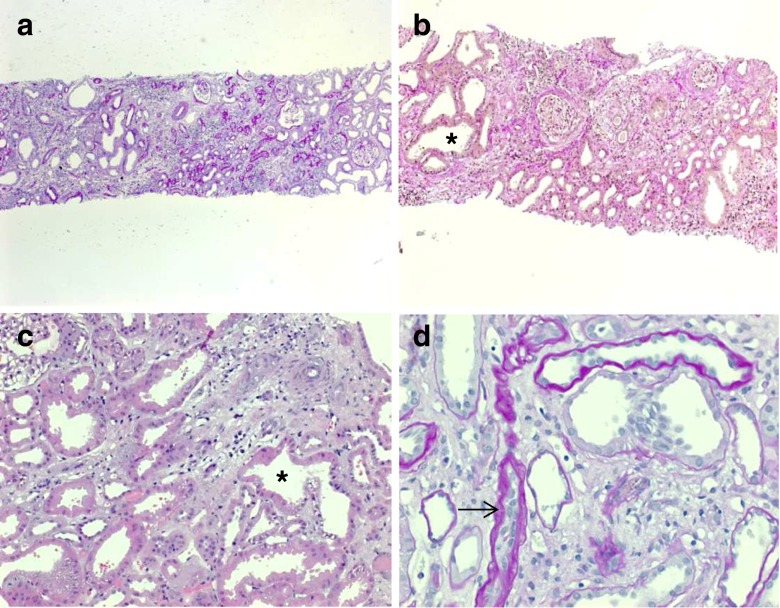
Figures
References
-
- Osathanondh V, Potter EL. Pathogenesis of polycystic kidneys. Survey of results of microdissection. Arch Pathol. 1964;77:510–512. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
