

Quantitative genome-wide genetic interaction screens reveal global epistatic relationships of protein complexes in Escherichia coli
- PMID: 24586182
- PMCID: PMC3930520
- DOI: 10.1371/journal.pgen.1004120
Quantitative genome-wide genetic interaction screens reveal global epistatic relationships of protein complexes in Escherichia coli
Abstract
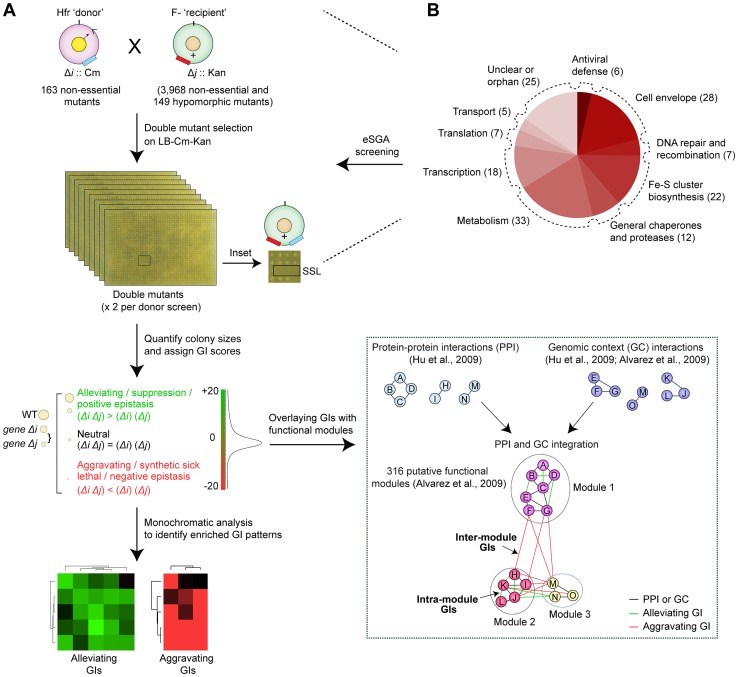
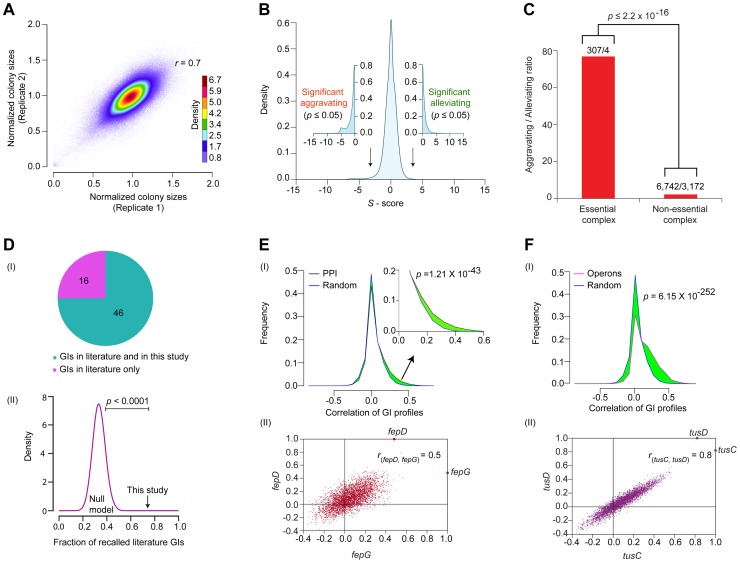
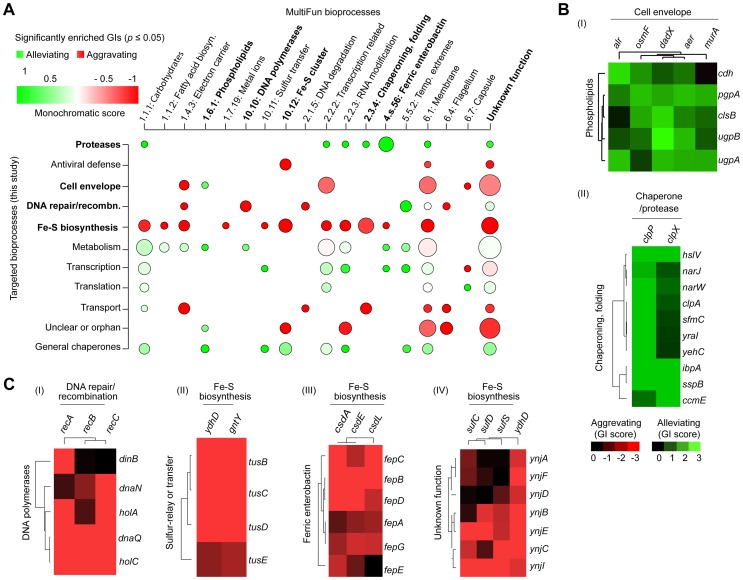
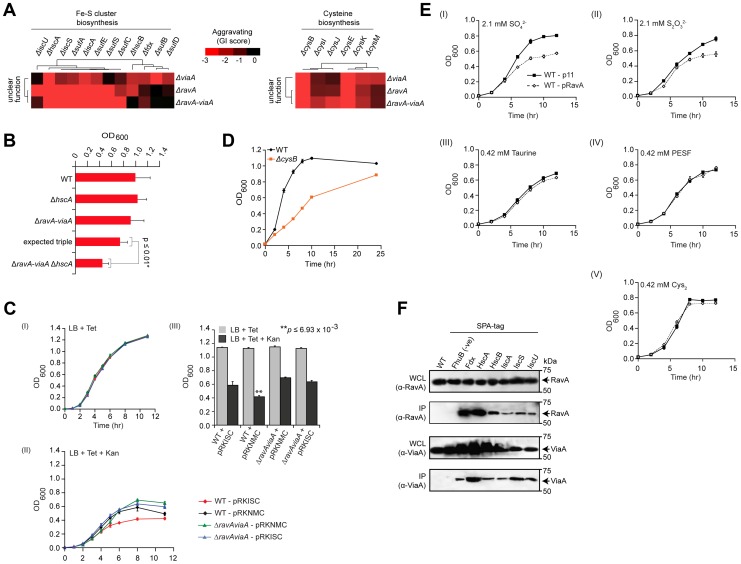
Large-scale proteomic analyses in Escherichia coli have documented the composition and physical relationships of multiprotein complexes, but not their functional organization into biological pathways and processes. Conversely, genetic interaction (GI) screens can provide insights into the biological role(s) of individual gene and higher order associations. Combining the information from both approaches should elucidate how complexes and pathways intersect functionally at a systems level. However, such integrative analysis has been hindered due to the lack of relevant GI data. Here we present a systematic, unbiased, and quantitative synthetic genetic array screen in E. coli describing the genetic dependencies and functional cross-talk among over 600,000 digenic mutant combinations. Combining this epistasis information with putative functional modules derived from previous proteomic data and genomic context-based methods revealed unexpected associations, including new components required for the biogenesis of iron-sulphur and ribosome integrity, and the interplay between molecular chaperones and proteases. We find that functionally-linked genes co-conserved among γ-proteobacteria are far more likely to have correlated GI profiles than genes with divergent patterns of evolution. Overall, examining bacterial GIs in the context of protein complexes provides avenues for a deeper mechanistic understanding of core microbial systems.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
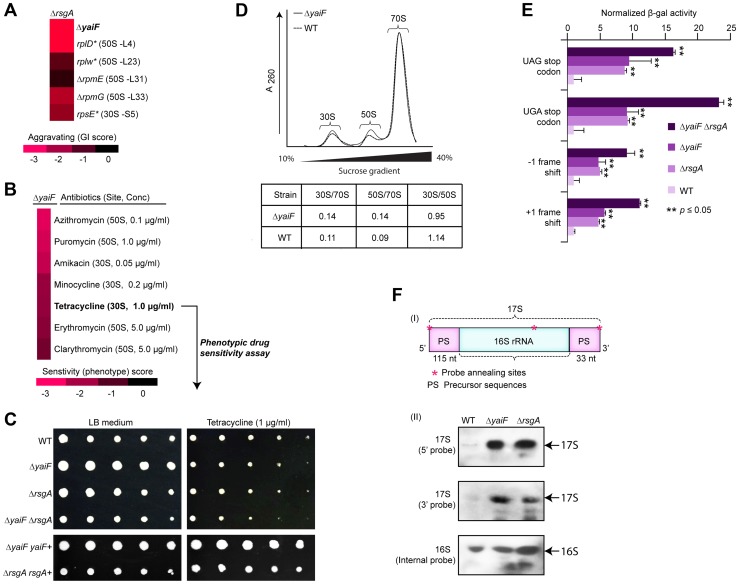
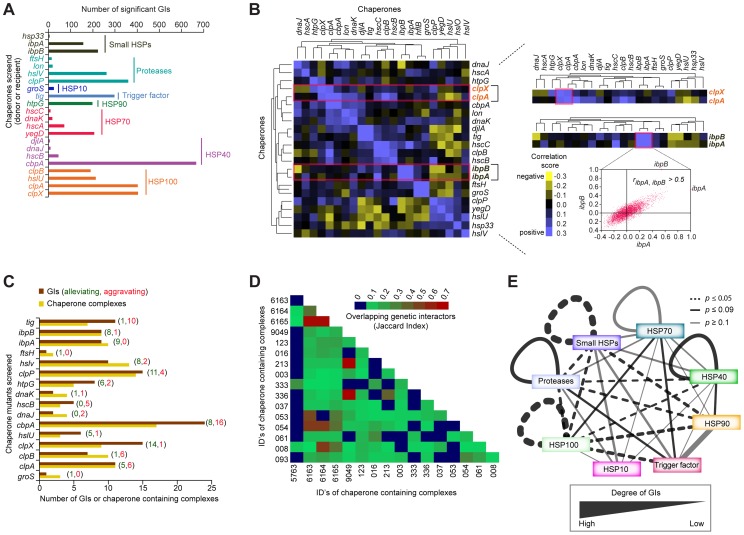
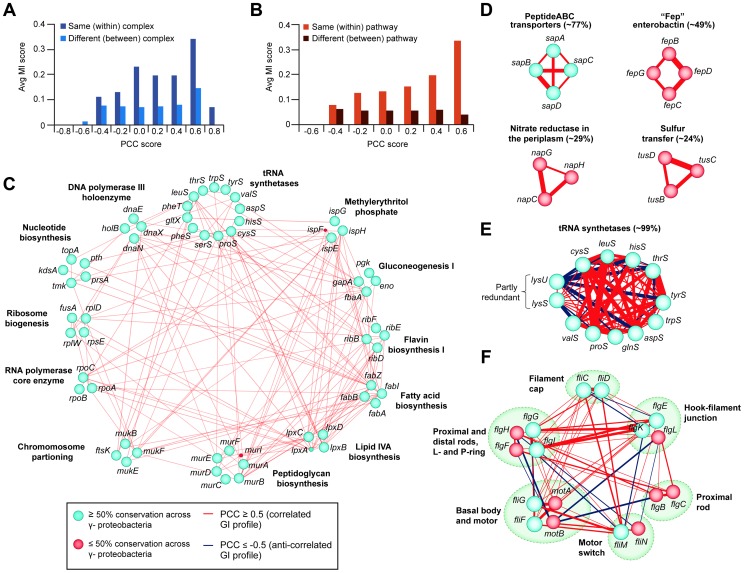
References
-
- Babu M, Musso G, Diaz-Mejia JJ, Butland G, Greenblatt JF, et al. (2009) Systems-level approaches for identifying and analyzing genetic interaction networks in Escherichia coli and extensions to other prokaryotes. Mol Biosyst 12: 1439–1455. - PubMed
-
- Moreno-Hagelsieb G, Collado-Vides J (2002) A powerful non-homology method for the prediction of operons in prokaryotes. Bioinformatics 18 Suppl 1: S329–336. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
