

Normalization of RNA-sequencing data from samples with varying mRNA levels
- PMID: 24586560
- PMCID: PMC3934880
- DOI: 10.1371/journal.pone.0089158
Normalization of RNA-sequencing data from samples with varying mRNA levels
Abstract
Methods for normalization of RNA-sequencing gene expression data commonly assume equal total expression between compared samples. In contrast, scenarios of global gene expression shifts are many and increasing. Here we compare the performance of three normalization methods when polyA(+) RNA content fluctuates significantly during zebrafish early developmental stages. As a benchmark we have used reverse transcription-quantitative PCR. The results show that reads per kilobase per million (RPKM) and trimmed mean of M-values (TMM) normalization systematically leads to biased gene expression estimates. Biological scaling normalization (BSN), designed to handle differences in total expression, showed improved accuracy compared to the two other methods in estimating transcript level dynamics. The results have implications for past and future studies using RNA-sequencing on samples with different levels of total or polyA(+) RNA.
Conflict of interest statement
Figures
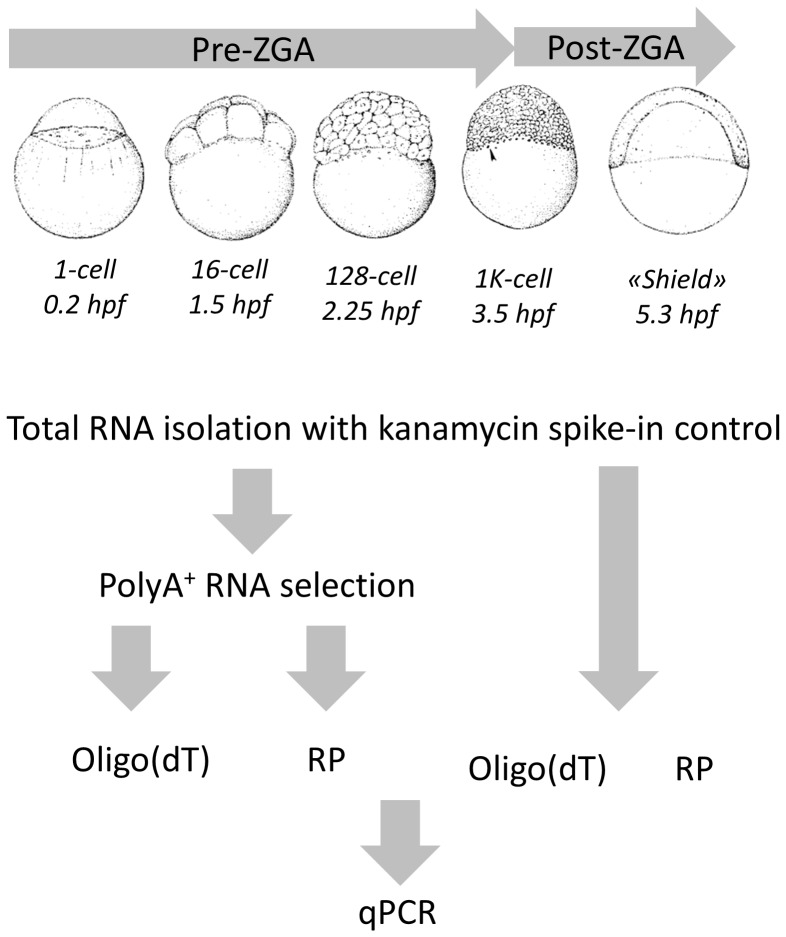


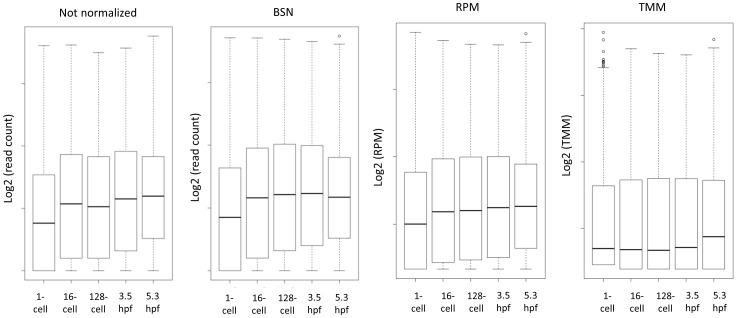
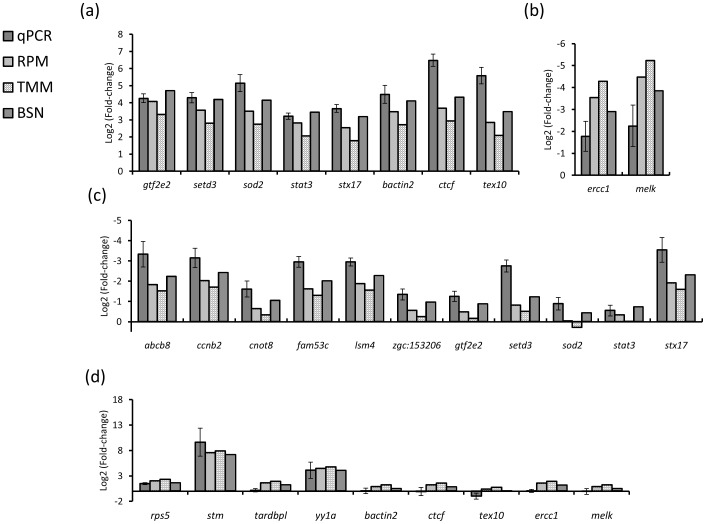
References
-
- Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, et al. (2012) A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform. bbs046 [pii];10.1093/bib/bbs046 [doi]. - PubMed
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628 nmeth.1226 [pii];10.1038/nmeth.1226 [doi]. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
