

DrugScorePPI knowledge-based potentials used as scoring and objective function in protein-protein docking
- PMID: 24586799
- PMCID: PMC3931789
- DOI: 10.1371/journal.pone.0089466
DrugScorePPI knowledge-based potentials used as scoring and objective function in protein-protein docking
Abstract
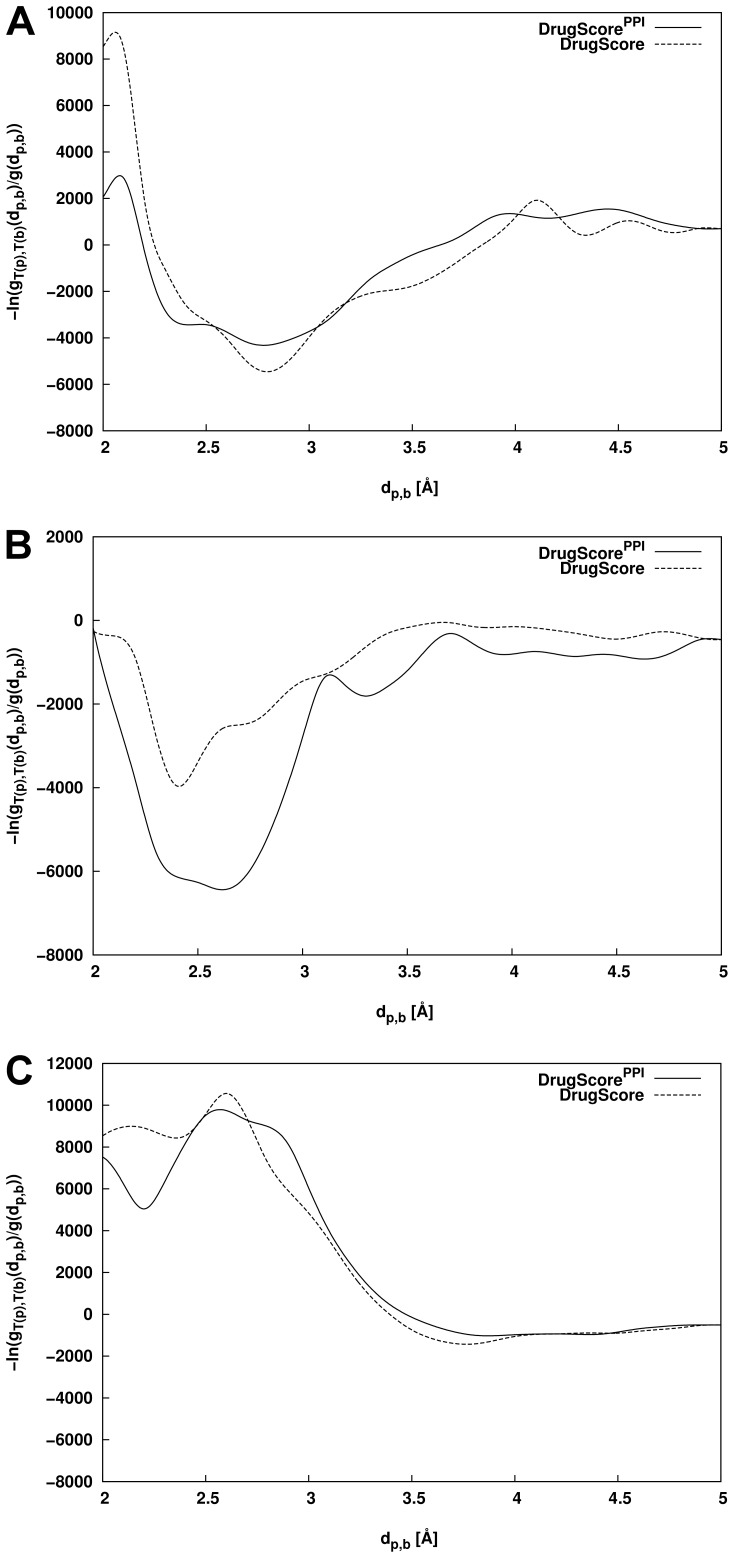
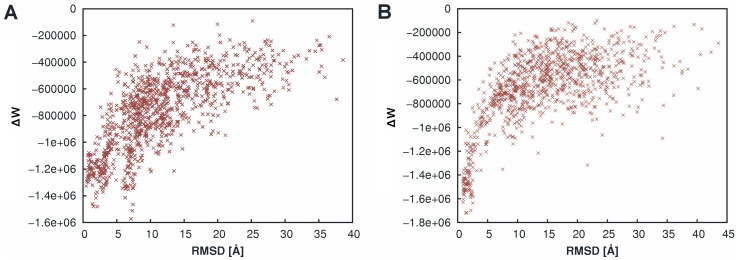
The distance-dependent knowledge-based DrugScore(PPI) potentials, previously developed for in silico alanine scanning and hot spot prediction on given structures of protein-protein complexes, are evaluated as a scoring and objective function for the structure prediction of protein-protein complexes. When applied for ranking "unbound perturbation" ("unbound docking") decoys generated by Baker and coworkers a 4-fold (1.5-fold) enrichment of acceptable docking solutions in the top ranks compared to a random selection is found. When applied as an objective function in FRODOCK for bound protein-protein docking on 97 complexes of the ZDOCK benchmark 3.0, DrugScore(PPI)/FRODOCK finds up to 10% (15%) more high accuracy solutions in the top 1 (top 10) predictions than the original FRODOCK implementation. When used as an objective function for global unbound protein-protein docking, fair docking success rates are obtained, which improve by ∼ 2-fold to 18% (58%) for an at least acceptable solution in the top 10 (top 100) predictions when performing knowledge-driven unbound docking. This suggests that DrugScore(PPI) balances well several different types of interactions important for protein-protein recognition. The results are discussed in view of the influence of crystal packing and the type of protein-protein complex docked. Finally, a simple criterion is provided with which to estimate a priori if unbound docking with DrugScore(PPI)/FRODOCK will be successful.
Conflict of interest statement
Figures
Similar articles
-
DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction.J Med Chem. 2005 Oct 6;48(20):6296-303. doi: 10.1021/jm050436v. J Med Chem. 2005. PMID: 16190756
-
CLUB-MARTINI: Selecting Favourable Interactions amongst Available Candidates, a Coarse-Grained Simulation Approach to Scoring Docking Decoys.PLoS One. 2016 May 11;11(5):e0155251. doi: 10.1371/journal.pone.0155251. eCollection 2016. PLoS One. 2016. PMID: 27166787 Free PMC article.
-
Converging a Knowledge-Based Scoring Function: DrugScore2018.J Chem Inf Model. 2019 Jan 28;59(1):509-521. doi: 10.1021/acs.jcim.8b00582. Epub 2018 Dec 18. J Chem Inf Model. 2019. PMID: 30513206
-
Predictive and Experimental Approaches for Elucidating Protein-Protein Interactions and Quaternary Structures.Int J Mol Sci. 2017 Dec 5;18(12):2623. doi: 10.3390/ijms18122623. Int J Mol Sci. 2017. PMID: 29206185 Free PMC article. Review.
-
Mapping, Structure and Modulation of PPI.Front Chem. 2021 Oct 7;9:718405. doi: 10.3389/fchem.2021.718405. eCollection 2021. Front Chem. 2021. PMID: 34692637 Free PMC article. Review.
Cited by
-
Improving the accuracy of high-throughput protein-protein affinity prediction may require better training data.BMC Bioinformatics. 2017 Mar 23;18(Suppl 5):102. doi: 10.1186/s12859-017-1533-z. BMC Bioinformatics. 2017. PMID: 28361672 Free PMC article.
-
Different combinations of atomic interactions predict protein-small molecule and protein-DNA/RNA affinities with similar accuracy.Proteins. 2015 Nov;83(11):2100-14. doi: 10.1002/prot.24928. Epub 2015 Sep 23. Proteins. 2015. PMID: 26370248 Free PMC article.
-
A comprehensive survey of scoring functions for protein docking models.BMC Bioinformatics. 2025 Jan 22;26(1):25. doi: 10.1186/s12859-024-05991-4. BMC Bioinformatics. 2025. PMID: 39844036 Free PMC article.
-
A novel strategy for molecular interfaces optimization: The case of Ferritin-Transferrin receptor interaction.Comput Struct Biotechnol J. 2020 Sep 24;18:2678-2686. doi: 10.1016/j.csbj.2020.09.020. eCollection 2020. Comput Struct Biotechnol J. 2020. PMID: 33101606 Free PMC article.
-
Interaction between TNF and BmooMP-Alpha-I, a Zinc Metalloprotease Derived from Bothrops moojeni Snake Venom, Promotes Direct Proteolysis of This Cytokine: Molecular Modeling and Docking at a Glance.Toxins (Basel). 2016 Jul 20;8(7):223. doi: 10.3390/toxins8070223. Toxins (Basel). 2016. PMID: 27447669 Free PMC article.
References
-
- Gonzalez-Ruiz D, Gohlke H (2006) Targeting protein-protein interactions with small molecules: challenges and perspectives for computational binding epitope detection and ligand finding. Curr Med Chem 13: 2607–2625. - PubMed
-
- Metz A, Ciglia E, Gohlke H (2012) Modulating protein-protein interactions: from structural determinants of binding to druggability prediction to application. Curr Pharm Des 18: 4630–4647. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
