

Blood platelets in the progression of Alzheimer's disease
- PMID: 24587388
- PMCID: PMC3938776
- DOI: 10.1371/journal.pone.0090523
Blood platelets in the progression of Alzheimer's disease
Abstract
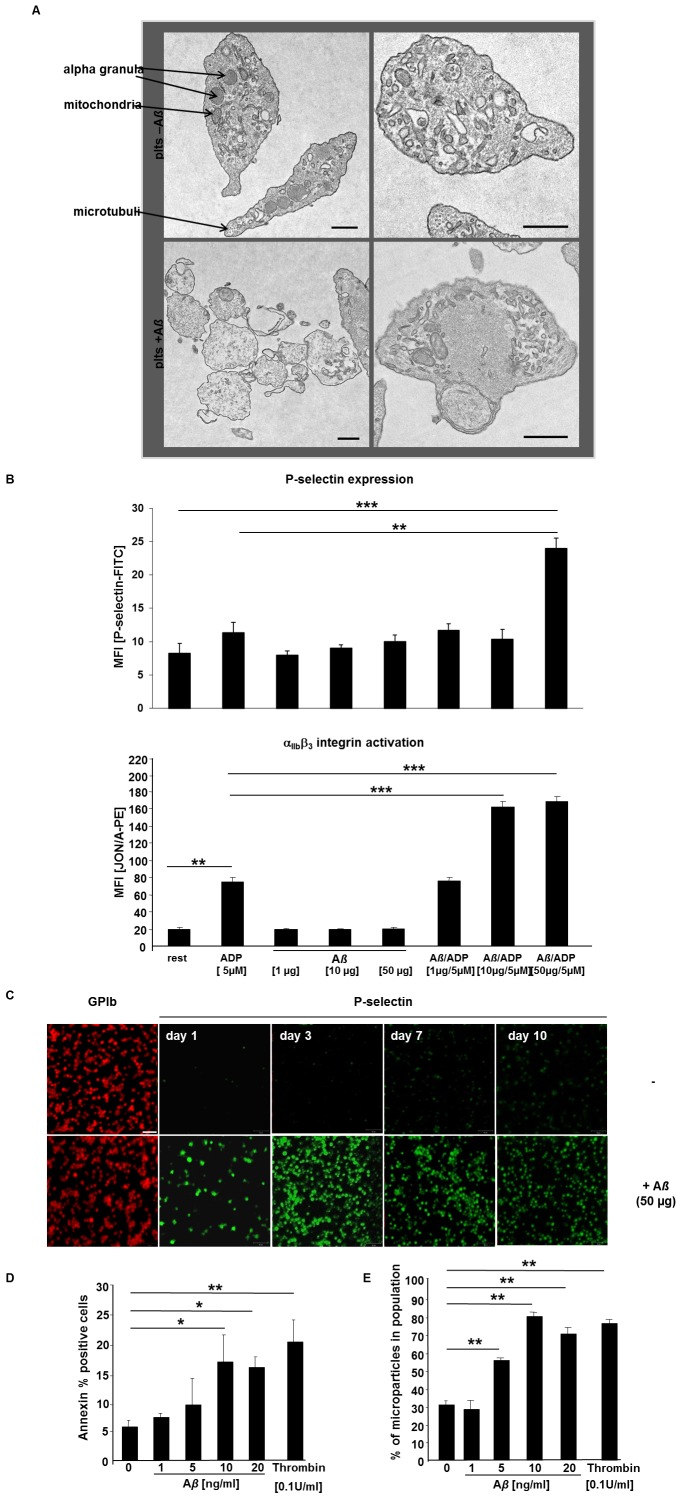
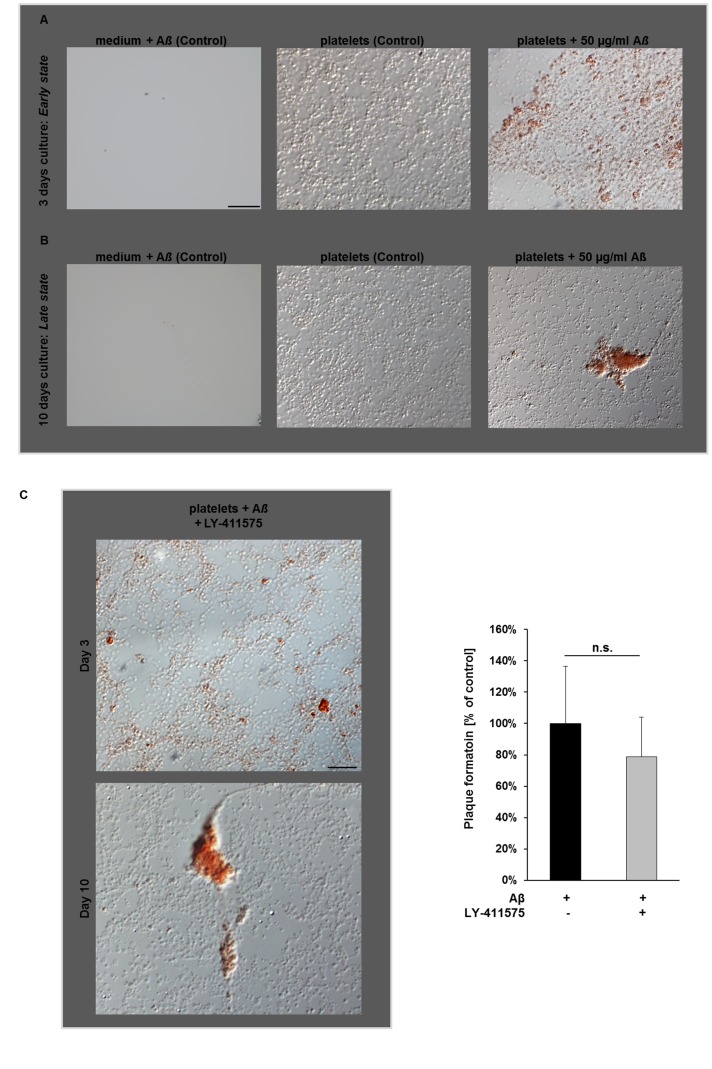
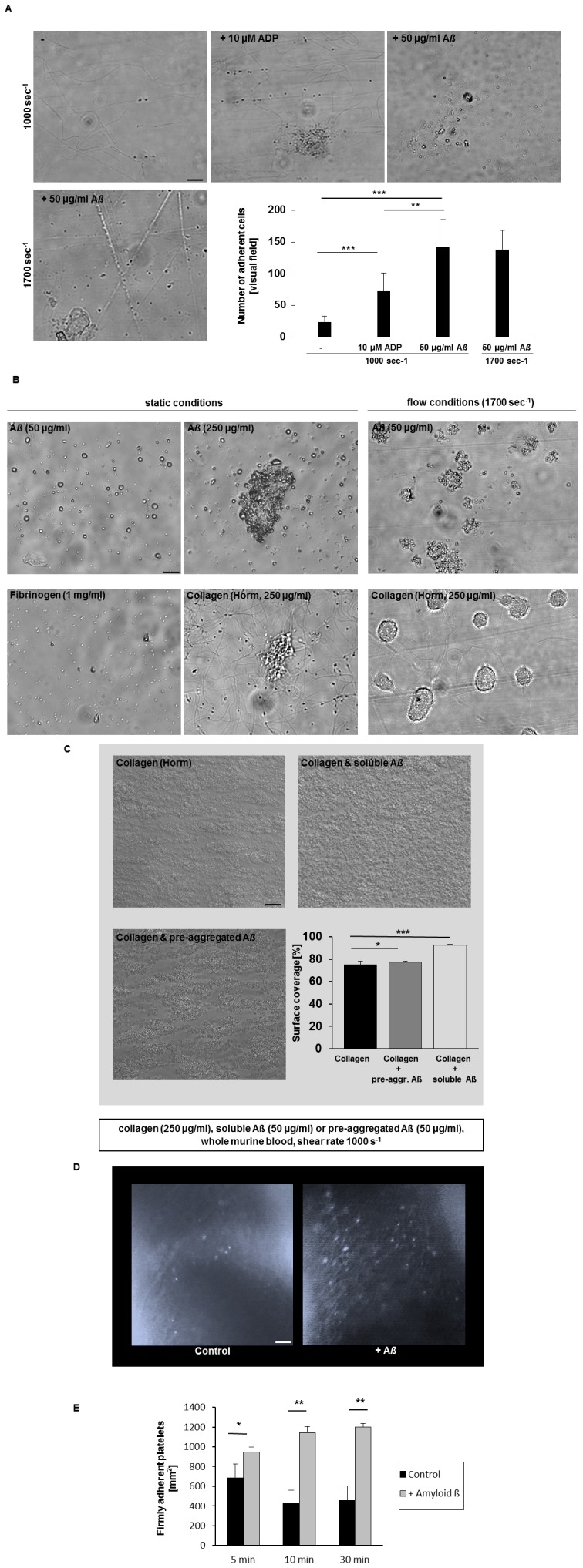
Alzheimer's disease (AD) is characterized by neurotoxic amyloid-ß plaque formation in brain parenchyma and cerebral blood vessels known as cerebral amyloid angiopathy (CAA). Besides CAA, AD is strongly related to vascular diseases such as stroke and atherosclerosis. Cerebrovascular dysfunction occurs in AD patients leading to alterations in blood flow that might play an important role in AD pathology with neuronal loss and memory deficits. Platelets are the major players in hemostasis and thrombosis, but are also involved in neuroinflammatory diseases like AD. For many years, platelets were accepted as peripheral model to study the pathophysiology of AD because platelets display the enzymatic activities to generate amyloid-ß (Aß) peptides. In addition, platelets are considered to be a biomarker for early diagnosis of AD. Effects of Aß peptides on platelets and the impact of platelets in the progression of AD remained, however, ill-defined. The present study explored the cellular mechanisms triggered by Aß in platelets. Treatment of platelets with Aß led to platelet activation and enhanced generation of reactive oxygen species (ROS) and membrane scrambling, suggesting enhanced platelet apoptosis. More important, platelets modulate soluble Aß into fibrillar structures that were absorbed by apoptotic but not vital platelets. This together with enhanced platelet adhesion under flow ex vivo and in vivo and platelet accumulation at amyloid deposits of cerebral vessels of AD transgenic mice suggested that platelets are major contributors of CAA inducing platelet thrombus formation at vascular amyloid plaques leading to vessel occlusion critical for cerebrovascular events like stroke.
Conflict of interest statement
Figures




References
-
- Thal DR, Griffin WS, de Vos RA, Ghebremedhin E (2008) Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol 115: 599–609. - PubMed
-
- Honig LS, Tang MX, Albert S, Costa R, Luchsinger J, et al. (2003) Stroke and the risk of Alzheimer disease. Arch Neurol 60: 1707–1712. - PubMed
-
- Mielke MM, Rosenberg PB, Tschanz J, Cook L, Corcoran C, et al. (2007) Vascular factors predict rate of progression in Alzheimer disease. Neurology 69: 1850–1858. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
