

Virtual high-throughput ligand screening
- PMID: 24590723
- PMCID: PMC4073479
- DOI: 10.1007/978-1-4939-0354-2_19
Virtual high-throughput ligand screening
Abstract
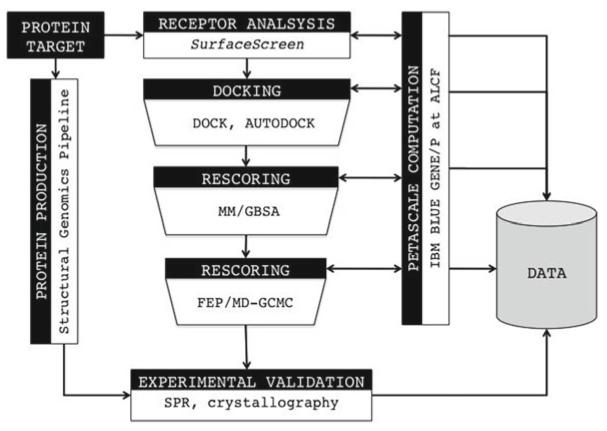
In Structural Genomics projects, virtual high-throughput ligand screening can be utilized to provide important functional details for newly determined protein structures. Using a variety of publicly available software tools, it is possible to computationally model, predict, and evaluate how different ligands interact with a given protein. At the Center for Structural Genomics of Infectious Diseases (CSGID) a series of protein analysis, docking and molecular dynamics software is scripted into a single hierarchical pipeline allowing for an exhaustive investigation of protein-ligand interactions. The ability to conduct accurate computational predictions of protein-ligand binding is a vital component in improving both the efficiency and economics of drug discovery. Computational simulations can minimize experimental efforts, the slowest and most cost prohibitive aspect of identifying new therapeutics.
Figures
References
-
- Macke T, Case DA. Modeling unusual nucleic acid structures. In: Molecular modeling of nucleic acids. American Chemical Society. 1998;682:379–393.
-
- Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
